Expressed Sequence Tag Analysis of the Erythrocytic Stage of Plasmodium berghei
Article information
Abstract
Rodent malaria parasites, such as Plasmodium berghei, are practical and useful model organisms for human malaria research because of their analogies to the human malaria in terms of structure, physiology, and life cycle. Exploiting the available genetic sequence information, we constructed a cDNA library from the erythrocytic stages of P. berghei and analyzed the expressed sequence tag (EST). A total of 10,040 ESTs were generated and assembled into 2,462 clusters. These EST clusters were compared against public protein databases and 48 putative new transcripts, most of which were hypothetical proteins with unknown function, were identified. Genes encoding ribosomal or membrane proteins and purine nucleotide phosphorylases were highly abundant clusters in P. berghei. Protein domain analyses and the Gene Ontology functional categorization revealed translation/protein folding, metabolism, protein degradation, and multiple family of variant antigens to be mainly prevalent. The presently-collected ESTs and its bioinformatic analysis will be useful resources to identify for drug target and vaccine candidates and validate gene predictions of P. berghei.
INTRODUCTION
The rodent malaria parasite Plasmodium berghei is similar to human malaria parasites, such as P. falciparum, in aspects of the structure, genome organization, physiology, and life cycle [1-3]. Therefore, P. berghei represents a practical and relevant model organism for experimental studies of malaria [4]. To improve the utility of models, such as P. berghei in the development of drug target and vaccine candidates for malaria, the genome sequence and actual transcripts are required as primary sources of biological information.
The genome of P. berghei is organized into 14 chromosomes, with an estimated genome size of 18 Mb [4]. Partial shotgun sequencing of the P. berghei genome and transcription profile analysis with genome survey sequences (GSS) is having significant contribution to many fields of malaria research [4]. However, most gene prediction of P. berghei have been based on bioinformatic analyses using computer software. However, the high A/T contents of the plasmodum genome, excluding P. vivax, hamper the prediction of the gene structure, resulting about 60% of the predicted genes encoding hypothetical proteins [5]. Therefore, it is necessary to verify the prediction with complementary DNA (cDNA), such as expressed sequenced tag (EST), a short contiguous subsequence of a transcribed DNA sequence, as a rapid means of gene identification to obtain useful information from a genome sequence, especially for intron-containing eukaryotes. Currently, large-scale random sequencing of ESTs is preceding concurrent with the plasmodum genome project [6-9]. Previous efforts to generate ESTs by random clones of a P. berghei cDNA library have accelerated the gene discovery processes [7]. The present study constructed a SMART™ PCR-amplified cDNA library from mixed blood stages of P. berghei parasites to enrich for full-length transcripts for detection of rare transcripts and transcript isoforms and determination of the relative abundance of transcripts. Here, we report the analysis of the P. berghei ESTs, including abundance, prevalence of protein domains, and functional categorization.
MATERIALS AND METHODS
Parasite collection
P. berghei ANKA strain (kindly provided by Dr. Eun-Taek Han, Department of Parasitology, Kangwon National University) was used to infect 6-week-old CL57B/6 mice. The blood stage of the parasite was used for cDNA library construction. Blood was collected by heart puncture under anesthesia and leukocytes were obtained using Plasmodipur leukocyte filters (Euro-Diagnostica, Malmö, Sweden). Parasites were released from their host RBCs by 0.15% saponin (0.5 volume of packed RBCs) (Sigma-Aldrich, St. Louis, Missouri, USA) in PBS, pH 7.5 (PBS) and agitated for 1-2 min until the suspension became a clear red color. The suspension was diluted by addition of 15 volumes of PBS, and the released parasites were collected by centrifugation [10].
Construction of P. berghei cDNA library
For construction of the P. berghei cDNA library, a PCR-based cDNA library was used with total RNA purified with TRIzol reagent (Gibco BRL, Rockville, Maryland, USA) following the instructions for the SMART cDNA library construction kit (BD-Clontech, Palo Alto, California, USA). cDNA was synthesized with a specially designed oligonucleotide (SMART IV) in the first-strand synthesis to generate high yields of full-length, double-stranded cDNA and 3' primer. Second-strand synthesis was performed by a long-distance PCR with Advantage 2 polymerase mix (Clontech). PCR products were extracted with phenol: choloroform (25:24) to remove the polymerases, digested with SfiI, and size-fractionated using a ChromaSpin-400 column (Clontech) to exclude cDNAs <500 bp. The cDNA mixture was ligated into the λ TriplEx2 vector (Clontech) and packaged using the GigaPack III Plus packaging extract (Stratagene, La Jolla, California, USA) according to the manufacturer's inst ructions.
In vivo excision and random sequencing
The titer and percentage of recombinant phages in the library was determined to 1×108 plaque forming units with 95% as recombinant clones. Escherichia coli strain BM25.8 cells were transduced with recombinant phage, from which the massive excision of the pTriplEx2 phagemid library was accomplished according to the manufacturer's instruction (Clontech). After in vivo excision, bacterial colonies were randomly selected and grown in LB-ampicillin broth by incubation with shaking at 31℃ overnight. Then, plasmids from selected colonies were extracted using the DNA-spin Plasmid DNA Purification Kit (iNtRON Biotechnology, Seoul, Republic of Korea) and sequenced with a PE377 DNA sequencer (Perkin-Elmer, Boston, Massachusetts, USA) using the Bigdye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, California, USA).
Bioinformatic analysis
The ESTs were initially analyzed with well-established procedure for EST sequence processing and annotated using the PESTAS automated EST analysis platform (http://pestas.kribb.re.kr) [11-13]. Each EST cluster was analyzed using BLASTX against the GenBank non-redundant protein database (April 2010 release) and Plasmodium annotated protein database in PlasmoDB (ver. 7.1, released November 2010, http://plasmodb.org/plasmo/) with an E-value of <10-5 for selection of matching [14]. After the first assignment, a BLASTN and TBLASTX search of the unmatched EST clusters was performed against the P. berghei EST and genome database in PlasmoDB to ascertain whether they were encoded in the P. berghei genome as putative new transcripts. EST cluster-associated GO terms were functionally classified based on protein-level annotation using BLAST2GO (cut-off ≤1e-10) [15]. Functional domains in novel clusters were assigned using InterProScan (HMMPfam, HMMSmart, HMMTigr, HMMPanther, and Superfamily, flagged as true by InterProScan with E-value <1e-2) [16]. All of the P. berghei ESTs generated from this study were submitted to the dbEST division of GenBank with accession numbers (HS576390-HS586433). Based on our ESTs, a specific P. berghei EST database (P. berghei EST DB) was constructed (http://parasite.knu.ac.kr).
RESULTS
P. berghei ESTs
The 12,000 clones containing DNA inserts were sequenced, sequence <100 bp were removed, and the remainder was processed with bioinformatic software programs to generate high quality ESTs. First, total ESTs were aligned against a non-redundant database of mouse gene for exclusion of mouse DNA contamination and 4 ESTs displayed encoding mouse genes (0.04%). A total of 10,040 ESTs having an average length of 643 bp and 74% [A+T] content were produced (Table 1). Cluster analysis with the processed ESTs, using TGICL, assembled 10,040 ESTs into 2,462 EST clusters with 1,432 contigs containing at least 2 or more overlapping sequences and 1,030 ESTs remained as singletons. The sequences of assembled contigs could be up to 2.5 kb in length and were composed of an average of 6.3 ESTs. The 2,462 EST clusters were compared with the P. berghei annotated protein database in PlasmoDB; 2,043 (83%) EST clusters were annotated with P. berghei proteins showing significant BLASTX matching at the cutoff value of <1e-5 with 419 EST clusters remaining unmatched. From BLASTX analysis, we found that 244 genes with predicted coding regions were fully covered by EST clusters.

Transcriptome features of Plasmodium berghei EST
After the first assignment, a BLASTN and TBLASTX search of the 419 unmatched EST clusters was performed against P. berghei EST databases in PlasmoDB. Of the 419 EST clusters, 371 (88.5%) displayed matching to P. berghei EST in the databases (Table 1). The 48 unmatched EST clusters were aligned using a BLASTN and TBLASTX analysis against a non-redundant protein database at the National Center for Biotechnology Information (NCBI) and the P. berghei genome database to ascertain whether they were encoded in the P. berghei genome. Corresponding sequences were apparent with 48 (11.5%) of these non-matched ESTs, most of them were hypothetical proteins with unknown function, implicating these EST clusters as putative new transcripts in P. berghei (Table 1). These results support the view that the P. berghei protein database remains incomplete.
Abundant P. berghei ESTs
We examined the redundancy of EST clusters, because redundant EST appears to reflect the highly expressed genes, which can highlight the importance of the genes in their respective biological pathways. The most abundantly detected transcripts (i.e., EST clusters containing more than 50 ESTs) are summarized in Fig. 1. Many of them corresponded to ribosomal, hypothetical, membrane proteins, or proteins in the purine salvage pathway.
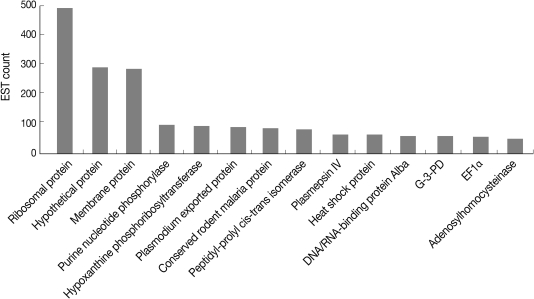
The abundant transcripts in Plasmodium berghei.
Protein domains in P. berghei ESTs
We further analyzed EST clusters with Pfam (http://pfam.sanger.ac.uk) to catalog the protein domains present in the P. berghei EST datasets, because the identification of domains that contain within proteins, especially hypothetical proteins, can provide insights into their functions [17]. The prevalence of protein domains in P. berghei ESTs is summarized in Table 2 showing RNA recognition motifs (RRM; PF00684), which contained the RNA binding protein implicated in regulation of splicing, RNA stability, and translation, to be most prevalent. Proteasome, subunit alpha/beta (PF00276), variant antigen Yir/Bir/Cir (PF01849) and chaperonin Cpn60/TCP-1 (PF00009) are among the top 10 Pfam families in the ESTs. These results together with previous results, the redundancy of EST clusters, indicate that proteins in the asexual blood stages of P. berghei are mainly related in translation/protein folding and degradation. Human malaria parasites evade the host immune response through the members of multigene families, such as Var, Rif, and Stevor, encoding virulence determinants of cytoadhesion and antigenic variation. In rodent malaria parasites (P. yoelii, P. berghei, and P. chabaudi), a large paralogous multigene family of variant antigens, Yir/Bir/Cir, is also conserved [18]. Consistent with their importance, variant antigen Yir/Bir/Cir (PF01849) displayed a significant portion in the prevalence of protein domains in P. berghei ESTs.

The prevalence of protein domains in P. berghei ESTs
Functional categorization of P. berghei ESTs
The EST clusters were grouped as functional categories based on GO molecular functions. GO, which consists of 3 major ontologies, i.e., biological process, molecular functions, and cellular components, is the most widely used method to predict gene families and functions of EST sequences. The BLAST2GO program was used in the functional classification of the P. berghei ESTs. From this, 1,631 (66.2%) EST clusters were assigned to biological processes (523; 21.2%), cellular components (377; 15.3%), and molecular functions (731; 29.7%) (Fig. 2). Consistent with our expectation, the majority of the genes with functional assignments were related to translation/protein folding, ribosomal structure, and metabolism. In particular, EST clusters were classified into proteolysis (GO: 0006508), including berghepain-2, a falcipain-2 homologue in P. berghei, plasmepsin, and many aminopeptidases has a significant proportion in biological processes. Interestingly, aminopeptidases, especially methionine aminopeptidases (MetAP), were more frequently detected compared with other proteinases, indicating the exuberant expression of MetAP in P. berghei. Aminopeptidases have been suggested as new targets for anti-malarial drug development [19]. In P. falciparum, 4 methionine aminopeptidases are expressed among the 9 identified aminopeptidases [20]. An inhibitory compound against MetAP, XC11 was active against both chloroquine sensitive and resistant P. falciparum 3D7 in culture and P. berghei in mice, implicating MetAP as an important drug target for anti-chloroquine resistant malaria [21].
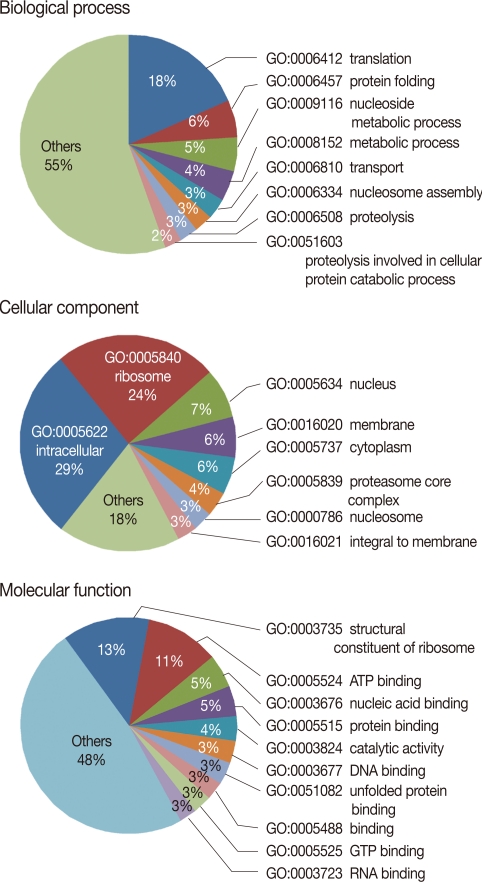
Gene ontology mapping for P. berghei EST clusters using BLAST2GO. The genes were functionally categorized based on the Gene Ontology Consortium. Level 3 of the assignment results are shown.
The substantial proportions of transport proteins (GO: 0006810, 3%) together with intracellular protein transport proteins (GO: 0006886, 1.5%, data not shown), and vesicle-mediated transport proteins (GO: 0016192, 1.3%, data not shown) were indicative of the importance of intracellular and extracellular trafficking of proteins in this pathogenic parasite. Moreover, as consistent with Fig. 1, chaperones (GO: 0031072) constituted a significant proportion in the class of molecular functions (1%, data not shown) and also chaperonin (PF00009) and HSP70 (PF01020) are frequently detected domains in P. berghei ESTs (Table 2). Therefore, these results suggested that protein trafficking is essential for survival of Plasmodium in blood stages and a promising drug target to combat against human malaria.
DISCUSSION
The availability of genome, transcriptome, and proteome data of Plasmodium spp. has greatly advanced the understanding of the biology of these organisms. However, the high A/T content in the P. berghei genome hampers prediction of open reading frames or identification of target genes. Therefore, this large EST collection can provide high quality data regarding coding sequences and expressed gene profiles. As the first expression profile analysis of P. berghei, 5,582 ESTs and 5,482 GSSs were functionally classified [7]. Thereafter, the transcription profile of asexual stage of P. berghei was analyzed by hybridization to a P. berghei GSSs amplicon DNA microarray categorizing into the 4 strategies of gene expression, such as housekeeping, host-related expression, strategy-specific expression, and stage-specific expression. In his study, 10,040 ESTs enriched in intact 5' ends from P. berghei cDNA library were assigned to functional categories based on GO using BLAST2GO revealed the expressed gene profile of P. berghei during asexual blood stages. The redundancy of EST clusters and the prevalence domain analysis of P. berghei proteins could provide clues for determining their functions and their importance in metabolic pathways. Among the highly abundant transcripts (Fig. 1), the enzymes engaged in nucleotide metabolism, purine nucleotide phosphorylase (PNP), and hypoxanthine phosphoribosyltransferase (HPRT) were well detected by multiple ESTs. Plasmodium spp. are unable to synthesize purine de novo and alternatively rely on the salvage pathway with host purines. Hypoxanthine, a primary source of purine, is produced by PNP or in human serum and converted into inosine monophosphate (IMP) by HPRT. Immunocillin-H, a PNP transition state analogue, inhibits P. falciparum growth by inhibiting PNP [22, 23]. The significant dependence on HPRT for nucleotide synthesis was presently reflected by its abundance in the ESTs. The results are consistent with the focus on HPRT as a promising drug target for the development of anti-malarial therapies, by virtue of its different characteristics from host protein [24].
The immunosuppressants FK506 and rapamycin have anti-malarial properties by virtue of binding to the target FK506-binding protein (FKBP) having peptidyl-prolyl cis-trans isomerase activity. However, their mechanisms of action against malaria parasites are unclear [25]. In P. falciparum, PfFKBP35 with peptidyl-prolyl cis-trans isomerase activity has been reported [26]. PfFKBP35 is inhibited by FK506, rapamycin, and calineurin, although the latter is independent of FK506 binding. The immunosuppressive peptide cyclosporin A also inhibits the growth of malaria parasites, presumably by binding to cyclophilins (distinct intracellular prptidyl-prolyl cis-trans isomerase) [27]. Peptidyl-prolyl cis-trans isomerase activity that is completely inhibited by cyclosporin A but not by FK506 or rapamycin has been detected in extracts of P. falciparum [27]. These results support the suggestion that P. falciparum probably contains more cyclophilins. Peptidyl-prolyl cis-trans isomerase differing from the PfFBPR35 homologue was highly abundant in the P. berghei ESTs (Fig. 1).
Similar with the abundance of heat shock protein (HSP) from P. vivax as evident from EST analysis [28], HSP constituted 1.34% of all P. berghei ESTs. HSP70 (0.8%), 1 of the 2 major HSPs (HSP90 and HSP70), was more abundant compared to HSP90 (0.3%) in the P. berghei library. The importance of HSP70 as a molecular chaperone concerning temperature changes between vector and host, and protein trafficking, has made the protein an important potential anti-malarial drug target. The semisynthetic Hsp90 inhibitor (17-[allylamino]-17-demethoxygeldanamycin) that is active against Plasmodium HSP90 is effective in attenuating parasite growth and prolonging survival in a mouse model of malaria [29].
Malaria parasites possess a relict plastid called the apicoplast that is homologous to the chloroplast of plants. The apicoplast contains the capacity for besides basic metabolic processes such as protein translation, and the biosynthesis of fatty acids, isoprenoids, iron-sulphur clusters and heam, which are essential for parasite survival. However, fewer than 50 proteins are encoded for in the apicoplast genome; the vast majority of metabolic pathway related proteins are encoded in the nuclear genome and are subsequently transported to the apicoplast [30, 31]. Interestingly, the transport machinery of apicoplast targeting proteins is similar with that in the chloroplast; the translocon of the outer envelope of chloroplast (TOC) and translocon of inner envelope of chloroplast (TIC) complexes are assumed to promote protein transport [32, 33]. Analysis of the presently obtained ESTs revealed 2 TIC components, Tic20 and Tic22, and no TOC components, consistent with a previous report [34]. Because the apicoplast is non-photosynthetic, sources of energy and carbon for such anabolic synthesis should be required. As an important cytosolic source of carbon, dihydroxyacetone phosphate (DHAP) is imported and converted to glycerol-3-phosphate (G3P), which is a precursor for phospholipids synthesis. G3P is sequentially acylated by glycerol-3-phosphate acyltransferase (ACT1) and 1-acyl-glycerol-3-phosphate acyltransferase (ACT2) to produce phosphatidic acid [35]. One enzyme in this pathway, ACT2, was found to be encoded for by the P. berghei ESTs.
In the present study, 10,040 ESTs from P. berghei cDNA library were generated, increasing the number of P. berghei sequence in public database. Moreover, the present screening method, which used ESTs enriched in genes with intact 5' ends, provided 244 genes with predicted coding regions fully covered by 254 EST clusters, showing a powerful means for confirmation of in silico annotation and identification of the target genes. Also, 48 putative new transcripts encoded in P. berghei genome that did not match any EST and annotated protein database of P. berghei were identified. However, many of the EST assemblies (Fig. 1) together with these putative new transcripts were assigned to the categories that encode hypothetical proteins with unknown functions, indicating that further studies are needed to define their functions in metabolic pathways. In addition, many EST clusters from this study are contained long 5' and 3' untranslated regions (UTRs). The information of these regions can be useful for understanding gene regulation of Plasmodium. The constructed a specific P. berghei EST database (http://parasite.knu.ac.kr) based on our ESTs and genetic resources will be helpful for bioinformatics analysis and identification of interested genes of Plasmodium.
The presently-collected ESTs will be a useful resource to validate gene predictions, and extend our understanding of the biology of Plasmodium spp., and screening for drug target and vaccine candidates.
ACKNOWLEDGEMENTS
We thank Dr. Eun-Taek Han, Department of Parasitology, Kangwon National University, for kindly providing P. berghei ANKA strain. This study was supported by grant 2009-0075049 from the Basic Research Program of the Korea Science & Engineering Foundation (KOSEF) and the Brain Korea 21 Project in 2011. We especially acknowledge the KOSEF program (System development for application of genomic sequence information) 2007-004269 funded by the Korean government (MEST).