The Role of PI3K/AKT Pathway and NADPH Oxidase 4 in Host ROS Manipulation by Toxoplasma gondii
Article information
Abstract
Dendritic cell is one of the first innate immune cell to encounter T. gondii after the parasite crosses the host intestinal epithelium. T. gondii requires intact DC as a carrier to infiltrate into host central nervous system (CNS) without being detected or eliminated by host defense system. The mechanism by which T. gondii avoids innate immune defense of host cell, especially in the dendritic cell is unknown. Therefore, we examined the role of host PI3K/AKT signaling pathway activation by T. gondii in dendritic cell. T. gondii infection or T. gondii excretory/secretory antigen (TgESA) treatment to the murine dendritic cell line DC2.4 induced AKT phosphorylation, and treatment of PI3K inhibitors effectively suppressed the T. gondii proliferation but had no effect on infection rate or invasion rate. Furthermore, it is found that T. gondii or TgESA can reduce H2O2-induced intracellular reactive oxygen species (ROS) as well as host endogenous ROS via PI3K/AKT pathway activation. While searching for the main source of the ROS, we found that NADPH oxidase 4 (NOX4) expression was controlled by T. gondii infection or TgESA treatment, which is in correlation with previous observation of the ROS reduction by identical treatments. These findings suggest that the manipulation of the host PI3K/AKT signaling pathway and NOX4 expression is an essential mechanism for the down-regulation of ROS, and therefore, for the survival and the proliferation of T. gondii.
INTRODUCTION
Toxoplasma gondii is an opportunistic protozoan that can infect most warm-blooded animals, regardless of human or animal. It is suspected that about one third of the world population are infected with T. gondii and causes recognizable and symptomatic disease in individuals infected in utero or with compromised immune systems. Congenital toxoplasmosis can lead to ophthalmic and neurological sequelae such as motor abnormalities, chorioretinitis, seizures, developmental delay and hydrocephalus, which is an important cause of infant morbidity and mortality [1]. T. gondii can be latent in tissues in the form of cysts when the environment surrounding T. gondii is not preferable for their growth, and the cyst can be often found in brain. The blood vessels of the brain are known to be protected by the Blood Brain Barrier (BBB), and this barrier makes foreign substance or pathogen, impossible to infiltrate into brain. Though, T. gondii has developed a Trojan horse strategy to sneak into the brain using dendritic cells as a Trojan horse [2–6].
Dendritic cells (DCs) are excellent antigen-presenting cells (APCs) and important in the host immune response among the variety of immune cells against T. gondii infection. The production of IL-12 from DCs is important to inhibit T. gondii proliferation, and detection of T. gondii profilin by TLR11 has a crucial role for the control of IL-12 [7]. In addition, the intracellular location of TLR11 is thought to be crucial for the detection of T. gondii profilin after phagocytosis [8]. But it should be noticed that T. gondii also developed a way to circumvent host immune responses. For example, MHC class II expression is significantly lowered after IFNγ stimulation to Toxoplasma-infected host cells. IFNγ-induced STAT1 activity is essential for the regulation of T. gondii growth, it also has been reported that T. gondii inhibits STAT1 by upregulating suppressor of cytokine signaling (SOCS) proteins [9]. It is thought that this signal suppression inhibits the immune response of DCs and thereby T. gondii gains benefits for growth to survive.
Previous studies have shown that T. gondii proliferation in macrophages is proportional to the reduction of ROS in host cells, suggesting host ROS reduction is an essential factor for T. gondii proliferation [10]. In addition, IFNγ-stimulated dendritic cells were reported to inhibit proliferation of T. gondii through the production of toxic oxygen metabolites, but NO production was not increased, suggesting that NO is not one of the toxic metabolites for inhibition of T. gondii growth [11]. For the ROS regulation, specific signaling activity of the host cell might be important for ROS regulation, and the possibilities have been discussed that PI3K/AKT signaling pathway and FOXO transcription factors are the major players for this event.
PI3K is a ubiquitously expressed enzyme that is responsible for the regulation of various intracellular processes, such as insulin-dependent cell growth, membrane trafficking, and endosome fusion. The serine/threonine protein kinase B (PKB)/AKT is one of the major downstream targets of PI3K and is a central player in growth regulation of cells. Phosphorylation at Ser473 and Thr308 activates the kinase activity of AKT, which regulates multiple cellular processes that increase metabolism, growth, and synthetic processes and suppress apoptosis [12]. AKT is dephosphorylated Ser473 by the Pleckstrin homology domain leucine-rich repeat protein phosphatases 1 and 2 (PHLPP1/2) [13]. AKT activation induces different cell survival mechanisms. AKT phosphorylates and inactivates the pro-apoptotic factors Bad and procaspase-9, as well as the Forkhead family of transcription factors that induce the expression of pro-apoptotic factors such as Fas ligand [14].
In addition to activation of AKT by growth factor, it is known that AKT can be activated by pathogen infection. These AKT activities not only inhibit the accumulation of autophagy protein LC3 around T. gondii but also inhibit the apoptosis of host cells and affect the survival of T. gondii [15–17].
In this study, it is revealed that T. gondii can induce host AKT activation and ROS suppression in dendritic cell. We also confirmed that host AKT activation is important for the T. gondii proliferation which is related with reduction of ROS in host cells. Activation of PI3K/AKT signal pathway by T. gondii is indispensable machinery for NOX4 expression inhibition and host ROS down-regulation and all these components together positively contribute to the parasite proliferation in dendritic cells.
MATERIALS AND METHODS
Host cell culture
Murine dendritic cell line, DC2.4 cell was cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS and 1% antibiotic–antimycotic reagents (all from Gibco, Grand Island, New York, USA).
Parasites
T. gondii RH strain that expresses transgenic green fluorescent protein (GFP-RH) were obtained from Dr. Yoshifumi Nishikawa (Obihiro University of Agriculture and Veterinary Medicine, Japan). Tachyzoites were maintained in human retinal pigment epithelial cell line, ARPE-19 (ATCC, Manassas, Virginia, USA). And incubated at 37°C and 5% CO2 for 2 to 3 days.
T. gondii ESA extraction
Purified tachyzoites (3×108) were incubated at 37°C for 1 hr under mild agitation in test tubes containing 1.0 ml Phosphate buffered saline (PBS). After centrifugation for 10 min at 13,000 rpm, the supernatant including T.gondii excretory/secretory antigen (TgESA) was saved and stored at −20°C until been used.
Western blot
DC2.4 cells were cultured in 60mm plates and stimulated with RH tachyzoites or TgESA 24 hr and then treated with specific PI3 kinase inhibitors LY294002 (LY 0.1, 1, 10 μM), Wortmannin (WORT 2, 20, 200 nM), GDC-0941 (GDC 2.5, 25, 250 nM), ZSTK474 (ZSTK 0.1, 1, 10 nM) or AKT inhibitor AKTi1/2 (AKTi 0.025, 0.25, 2.5 μM). The protein samples were separated by SDS-PAGE gel and transferred onto polyvinylidene difluoride (PVDF) membranes and probed with the relevant antibodies. The protein bands were visualized by an ECL chemiluminescence kit (Amersham Biosciences, Freiburg, Germany) according to the manufacturer’s instructions. The bands were scanned and quantified using an imaging densitometer (Bio-Rad Laboratories, Inc., Hercules, California, USA). The primary antibodies used were rabbit anti-phosphor-AKT (Ser473) (#9271), AKT (#2966) (Cell Signaling Technology, Danvers, Massachusetts, USA), and rabbit anti-NOX4 (SC-30141), mouse anti-α-tubulin (SC-32293) (Santa Cruz Biotechnology, Dallas, Texas, USA).
Host gene expression analysis by reverse transcription-PCR (RT-PCR) and quantitative real-time reverse transcription-PCR (qRT-PCR)
After extract total mRNA with Trizol reagent (Invitrogen Life Technologies, Carlsbad, California, USA), the cDNA was synthesized using the Primer Script RT reagent kit (Takara Bio, Inc., Otsu, Japan) with 1 μg of total mRNA. Real-time PCR was performed using the SYBR-Premix Ex TaqTM II (2X) mix (Takara Bio, Inc., Otsu, Japan) with the Rotor-Gene Q Real Time PCR Systems (Qiagen, Hilden, Germany). The thermal cycle parameters were 95°C for 10 min and 40 cycles of each for 5 sec at 95°C and 40 sec at 60°C. Ct values for specific genes were corrected by the Ct value for the GAPDH housekeeping gene and expresses as ΔCt. Data represent mean±S.D., values of ΔCt. The primer sequences used were designed as follows, qmNOX1 F: CCA ATG TGG GAC AAT GAG TTT TC, R: AAC CCC CAC CGC AGA CTT; qmNOX2 F: CCC AAC TGG GAT AAC GAG TTC A, R: AGG GCC ACA CAG GAA AAC G; qmNOX3 F: TCT CCG GCT GCA CAA TGT C R: CTG CCT GCC ATT CAG CAT AG; qmNOX4 F: GCC TTT ATT GTG CGG AGA GAC T, R: TGA GAT GAT GGT GAC AGG TTT GT; qmDUOX1 F: GGG ATT CCC AGT CAT AAC TCA AGT, R: GCT CTC GAC TCC AGT AGC TGT TG; qmDUOX2 F: GGT CCT TGA CAA ACG GTT CAC, R: GGT GCG ACG TCC ACT GTT G; qmActin F: TCA TGA AGT GTG ACG TTG ACA TCC GT, R: CCT AGA AGC ATT TGC GGT GCA CGA TG. The primers used for RT-PCR were designed as follows, mNOX4 F: TGT TGC ATG TTT CAG GTG GT, R: TAC TGG CCA GGT CTT GCT TT; mβ-actin F: TGT TAC CAA CTG GGA CGA CA, R: TCT CAG CTG TGG TGG TGA AG.
Proliferation of T. gondii
DC2.4 cells were cultured in 12-well plates with glass coverslips and serum-starved for 4 hr before infected with T. gondii (GFP-RH) at a multiplicity of infection (MOI) of 1 or 5. For observation of inhibitor effect, sets of cells were pretreated with PI3Kinase inhibitors or AKT inhibitor for 2 hr, washed 1 time with PBS, and then infected with T. gondii. In order to “synchronize” the T. gondii replication starting point and exclude the possible interference from uninfected, free parasites, we have designed very narrow window for T. gondii infection (2 hr infection, removal of uninfected, free parasites from the media then incubation for 22 hr).
After 24 hr of infection, the coverslips were washed with PBS and then fixed with 4% formaldehyde. Cells were stained with Texas Red®-X phalloidin (Life Technologies Corporation, California, USA) to label F-actin and mounted with mounting solution with DAPI (Vector Laboratoies, Burlingame, California, USA) to stain nucleus. Cells were then imaged using confocal laser scanning microscope (Keica TCS SP5, Wetzlar, Hesse-Darmstadt, Germany). 100–200 cells were randomly selected in each preparation, and parasite replication was monitored by counting the number of tachyzoites per parasitophorous vacuole (PV). Three separate experiments were performed for statistical analysis of the results.
Invasion and attachment assay
Red/green invasion and attachment assays using indirect immunofluorescence were performed as described with the minor changes [18]. After brief fixation, external (attached) parasites were stained with T. gondii surface antigen SAG1 antibody (TP3, ab8313, Abcam, Cambridge, UK) before permeabilization with 3% BSA in PBS containing 0.1% TritonX-100. Internal (invaded) parasites were detected with anti-GFP antibody (sc-9996, Santa Cruz Biotechnology, Dallas, TX, USA), which recognizes internally expressing eGFP in T. gondii. Secondary antibodies were mixture of Alexa 488-cojugated goat anti-rabbit IgG and Alexa 594-conjugated goat anti-mouse IgG (Invitrogen, Co Dublin, Ireland). After 3 times washing by PBST, cells were mounted with mounting solution with DAPI to stain nucleus. The fluorescent images were acquired using a fluorescence microscopy.
Measurement of intracellular ROS
DC2.4 cells were cultured in 12-well plates containing glass coverslips and serum-starved for 4 hr before infection with T. gondii or treatment with TgESA for 24 hr. During the final 30 min, cells were exposed to 100 μM H2O2. Following washing with PBS, cells were incubated with 10 μM dihydroethidium (DHE) or 2′, 7′–dichlorofluorescin diacetate (DCFDA) for 30 min at 37°C in the dark. The cells image was obtained by an Olympus BX-51 florescence microscope or analyzed on a FACScan device (BD Biosciences, San Diego, California, USA).
Statistical analysis
The unpaired student’s t-test was used for statistical analysis. Three independent experiments data are presented as means± standard deviation. P values less than 0.05 are considered as been statistically significant.
RESULTS
T. gondii infection induced host AKT phosphorylation via PI3K in DC2.4 cell
In previous studies, it was reported that the AKT activity of host cells by T. gondii infection can inhibit its apoptosis [16]. To identify intracellular signaling pathway of the host cell which is modulated by T. gondii for its proliferation, we examined whether T. gondii infection can induce AKT phosphorylation (S473) in DC2.4 cells using immunoblot analysis. In addition, the effects of T. gondii ESA (TgESA) on host AKT were analyzed because, it is well known that TgESA plays an essential function to provide appropriate environment for invading the parasite into host cell [19].
To minimize the basal activity of AKT, DC2.4 cell was incubated in FBS-free RPMI1640 media for 4 hr before infection experiment. After pre-starvation, live T. gondii or TgESA were infected or treated, respectively for 24 hr, then proteins were extracted from the cells and western blotting was performed to assess host AKT activation by T. gondii. The phosphorylation levels of AKT (S473) in the infected and uninfected samples were determined by densitometric analysis and are shown in (Fig. 1A.) T. gondii infected or TgESA treated DC2.4 cells resulted in an induction of host AKT phosphorylation and the phosphorylation levels were increased obviously in T. gondii MOI- or TgESA concentration-dependent manner.

T. gondii-infection induced host AKT phosphorylation via PI3K in DC2.4 cell. (A) Live T. gondii tachyzoites (GFP-RH) or TgESA were challenged to DC2.4 cell for 24 hr at the indicated multiplicities of infection (MOI) or concentration, respectively, cell lysis solutions were used to probe for phosphor-Ser473 AKT and total AKT by western blot. (B, C) DC2.4 cells were infected with T. gondii at MOI5 for 24 hr, during the final 2 hr, PI3K inhibitors (LY, WM, GDC, ZSTK) or AKT inhibitor (AKTi) were treated to the cells. The protein levels of phosphor-Ser473 AKT and total AKT were also measured by western blot. Values represent the Mean±SD of triplicates. *P<0.05, **P<0.01, ***P<0.001, paired student t-test. Data are representative of 3 independent experiments. (LY: LY294002, WM: Wortmannin, GDC: GDC-0941, ZSTK: ZSTK474, AKTi: AKTi 1/2).
While screening for inhibitors which blocks T. gondii induced AKT phosphorylation, we found that higher concentration of LY294002 (10 μM) or wortmannin (200 nM) led to inhibition of the T. gondii infection-induced AKT phosphorylation in DC2.4 cells (Fig. 1B). Although wortmannin and LY294002 are well-known for their inhibiting effect on PI3K, there is an argument that they are not specific to PI3K and can inhibit mTOR pathway. Therefore, in order to confirm that the T. gondii induced AKT phosphorylation is regulated by PI3K in DC2.4 cells, the specific PI3K inhibitors, GDC-0941 and ZSTK474 recruited [20]. As a result, it is observed that GDC-0941 (250 nM) and ZSTK474 (10 nM) successfully inhibited T. gondii-induced AKT phosphorylation (Fig. 1B). A chemical reagent that is known as AKT specific inhibitor, AKTi 1/2, also clearly inhibited the T. gondii-induced AKT phosphorylation (Fig. 1C). These data suggested that T. gondii infection can activate host AKT via PI3K activation in DC2.4 cell.
PI3K inhibitors or AKT inhibitor inhibits T. gondii proliferation but not infection or invasion
The growth of parasite, highly relies on the host environment. We wondered whether T. gondii-induced PI3K-AKT activation has any cellular effect on host intracellular environment control and, thereby, T. gondii infection or growth efficiency. Therefore, the effect of PI3K /AKT activity on T. gondii proliferation in DC2.4 cells were monitored using fluorescent microscopy (Fig. 2A). As a result, it is observed that in control group (without inhibitor pretreatment), most tachyzoites divided 2 times to generate 4 parasites per PV. But when LY294002 or Wortmanin were treated, T. gondii proliferation was inhibited that the number of PV with 4 tachyzoites was approximately 50% less than that of untreated control group. GDC-0941 and especially, ZSTK474, newly developed PI3K-specific inhibitors, also showed suppression effect on T. gondii proliferation. AKTi also showed similar inhibitory effect for T. gondii growth.

PI3K/AKT inhibitors can suppress T. gondii proliferation but not its infection or invasion rate. (A) DC2.4 cells were pretreated with PI3K or AKT inhibitors for 2 hr before T. gondii infection (M1) for 24 hr. The number of tachyzoites in each parasitophorous vacuole (PV) in the host cells was counted under a fluorescence microscope. Scale bar=10 μm. (B) Invasion and attachment assay. Scale bar=20 μm. (C) T. gondii infection rate was measured by flow cytometry. (D) T. gondii proliferation was analyzed on a FACScan. Data were representative of 3 independent experiments.
Simultaneously, we questioned whether PI3K inhibitors can suppress T. gondii infection to host cells. In order to answer the question, using microscopic image data, T. gondii infection rate (infected cell/total cell in 24 hr infection) was calculated. As a result, PI3K inhibition had no significant effect on T. gondii infection to host (Fig. 2A). The effect of PI3K inhibitor on the invasion rate of T. gondii was also examined using Invasion and Attachment Assay which allows direct monitoring of parasite attachment and entry into host cells (Fig. 2B), which discriminates extracellular tachyzoites with SAG1 coat on from intracellular tachyzoites without SAG1 coat. As expected, we found that PI3K inhibitor treatment had no effect on parasite invasion, attachment and infection rate (1 hr) in DC2.4. Therefore, host PI3K pathway activation by T. gondii is indispensable for its proliferation but the pathway is not involved in initial stages of infection, attachment and invasion.
In addition, we tested whether the PI3K-specific or AKT inhibitors did not affect the initial infection rate through FACS. And the result showed that T. gondii infection rate was unchanged with both PI3K-specific or AKT inhibitor treatment (Fig. 2C). Like previous results, PI3K-specific or AKT inhibitor treatment suppressed T. gondii proliferation in DC2.4 cell (Fig. 2D). These results, again, confirmed that host PI3K and AKT activities are essential for T. gondii proliferation but not for invasion, attachment and infection rate.
T. gondii infection or TgESA treatment decreased H2O2-induced ROS level as well as intracellular ROS level via PI3K-AKT pathway in DC2.4 cell
Previously, proliferation of T. gondii within macrophages has been reported to be associated with reduced reactive oxygen species (ROS) in host cells [13]. Thus, it was questioned whether the activated PI3K/AKT signaling pathway of DC2.4 cells infected with T. gondii affected the production of ROS in host cells. Before studying the correlation between ROS and PI3K/AKT activity, we investigated whether T. gondii can reduce the intracellular ROS of DC2.4. DC2.4 cells were treated with TgESA or infected with T. gondii for 24 hr, respectively, and then, fluorescence of DCF-DA, a ROS probe, was measured by fluorescence microscopy or FACS (Fig. 3A, B). As a result, T. gondii-infected or TgESA-treated DC2.4 cells showed significantly reduced fluorescence, in other word, reduction of ROS. Next, it was tested whether the PI3K-specific inhibitor or AKT inhibitor could restore endogenous ROS reduced by TgESA treatment. As a result, reduced endogenous ROS by TgESA treatment was restored to basal level by PI3K inhibitor or AKT inhibitor treatment (Fig. 3C, D). It was also questioned whether TgESA treatment could reduce the ROS of host cells increased by exogenous ROS treatment. DC2.4 cells were pre-treated with TgESA for 24 hr and while incubation, PI3K-specific inhibitor or AKT inhibitor were added to media at 22 hr, and as an oxidation stress inducer, H2O2 was added at designated the time point (30 min before end of incubation). As a result, it is found that TgESA has capability to prevent the increase of intracellular hydrogen peroxide and superoxide level even in the presence of H2O2, and as expected the TgESA effect was nullified by PI3K-specific inhibitor or AKT inhibitor (Fig. 3E, F). These results suggest that TgESA treatment can reduces exogenous ROS as well as endogenous ROS of host cells via PI3K/AKT signaling pathway.
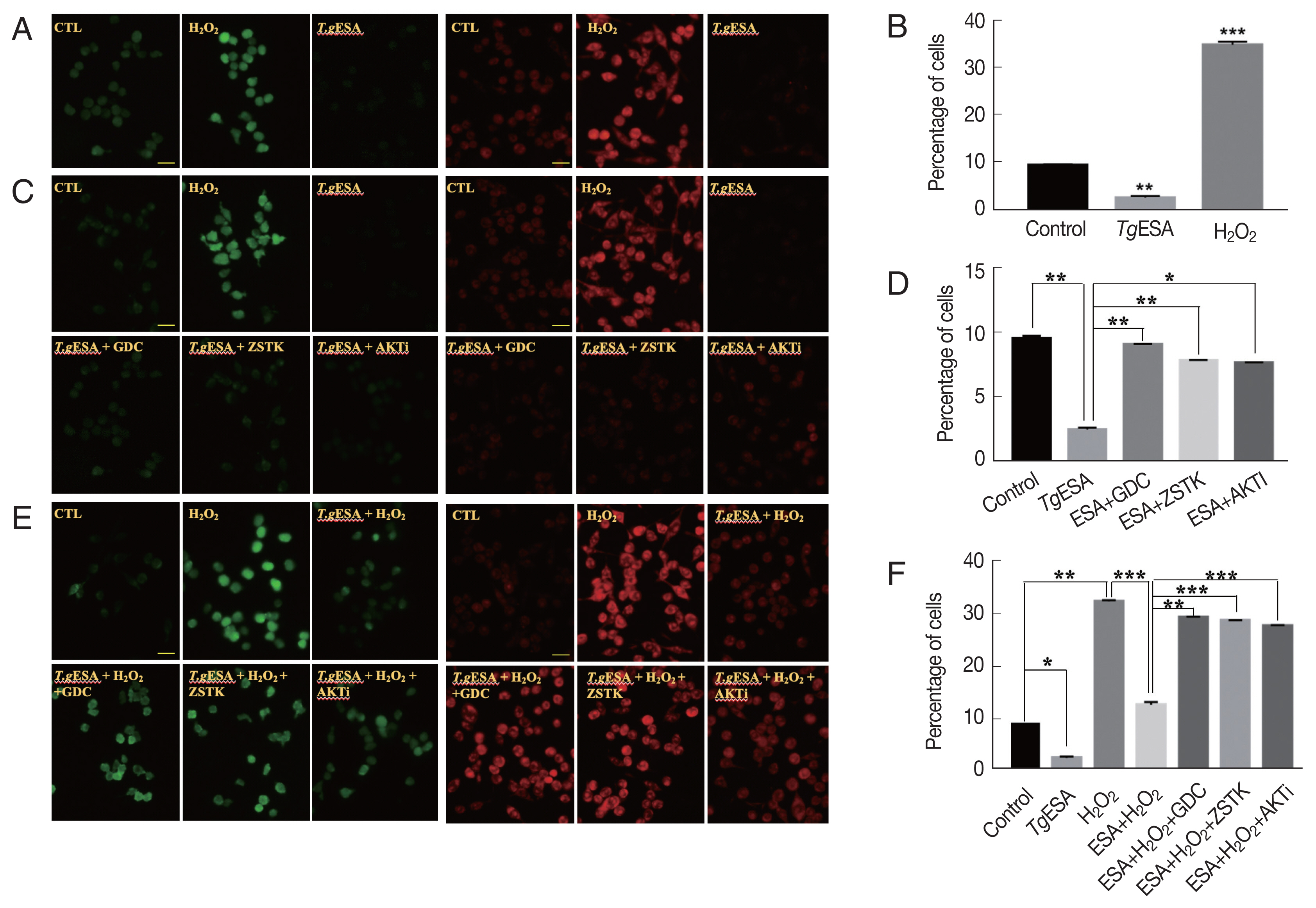
T. gondii infection or TgESA treatment diminished intracellular ROS generation as well as H2O2-induced ROS level via PI3K-AKT pathway in DC2.4 cell. (A) The intracellular ROS production levels were detected by fluorescence microscopy with DCFDA and DHE. (B) Flow cytometry analysis of intracellular ROS generation changes after TgESA treatment with DCFDA. (C) The effects of PI3K/AKT inhibitors on intracellular ROS generation were measured by fluorescence microscopy. (D) Flow cytometry analysis was performed to confirmed the effect of PI3K/AKT signaling activity on host ROS production with DCFDA. (E) The effects of PI3K/AKT inhibitors on H2O2-induced ROS level were measured by fluorescence microscopy. (F) Flow cytometry analysis was performed to confirmed the effect of PI3K/AKT signaling activity on H2O2-induced ROS level with DCFDA. Results shown are representative of 3 independent experiments. Scale bar=20 μm.
T. gondii infection to DC2.4 cell significantly down-regulated host NADPH oxidase(s) level via PI3K/AKT signaling pathway
Since NADPH oxidase is a major source of ROS production in cells, it is presumed that the T. gondii induced host PI3K/AKT activation may have an inhibitory effect on the NADPH oxidase expression or activation.
In order to test the hypothesis, mRNA expression of NADPH oxidase family in DC2.4 cells was monitored by real-time PCR method after T. gondii infection. As a result, it is found that only NOX4 in the NADPH family was significantly reduced by T. gondii infection (Fig. 4A). Indeed, NOX4 protein level in DC2.4 was dramatically decreased by T. gondii infection or TgESA treatment in accordance with NOX4 mRNA expression level change (Fig. 4B). To confirm whether the reduction of NOX4 is modulated by PI3K/AKT signaling pathway, the effect of PI3K/AKT inhibitors on NOX4 expression was tested using real-time PCR and Western blot analysis. As a result, it is observed that the reduction of host NOX4 level was reversed by inhibition of PI3K//AKT signaling pathway (Fig. 4C, D). In summary, T. gondii infection in dendritic cells induce activity of PI3K/AKT signaling pathway, and therefore, NOX4 expression will be reduced. This event might result in a reduction of ROS levels in the cell, and thus create a host environment that would favor the T. gondii proliferation (Fig. 5).
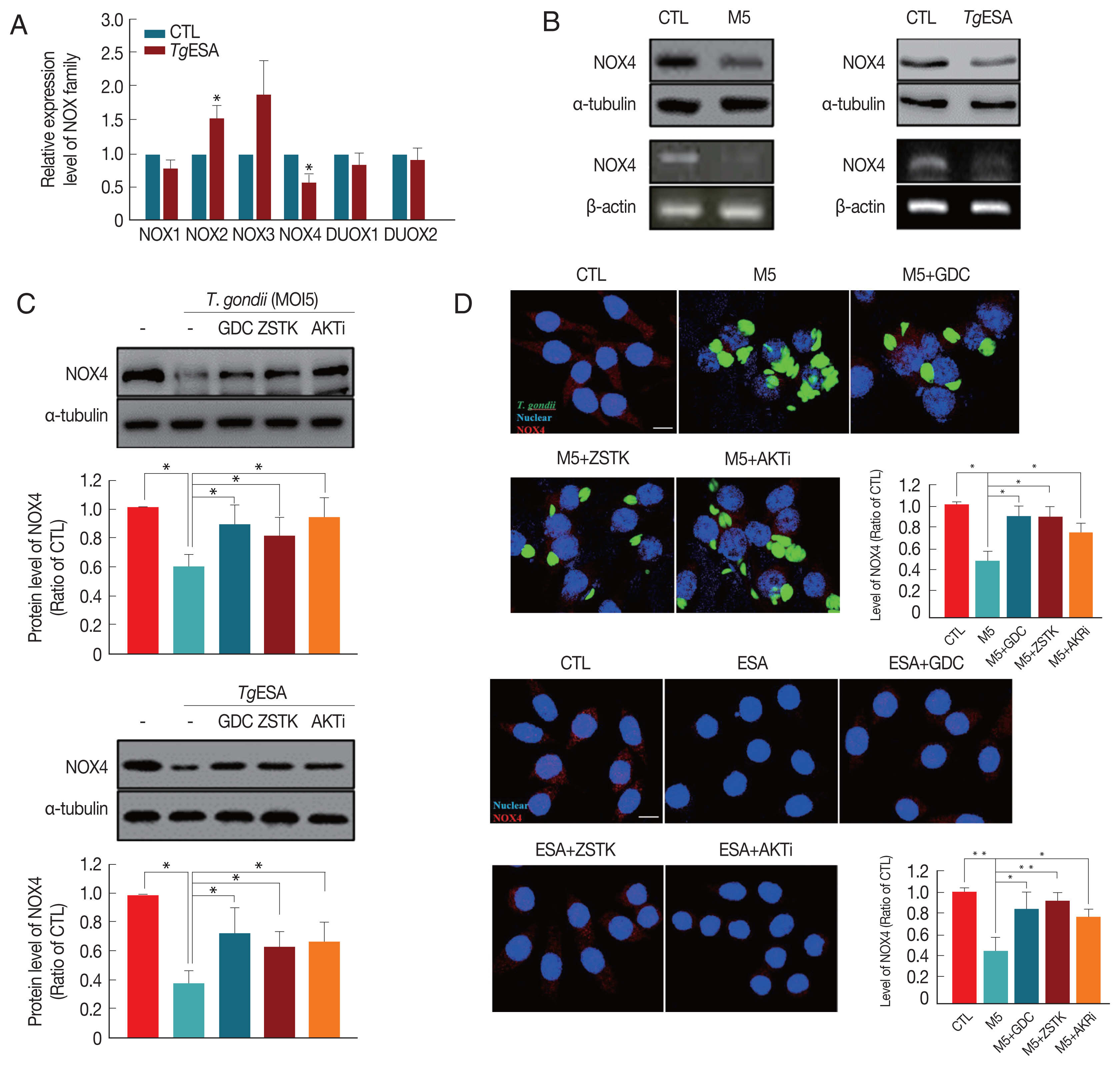
T. gondii infection significantly reduced host NADPH oxidase(s) level via PI3K/AKT signaling pathway. (A) DC2.4 cells were treated with TgESP and then the expression of NOX family was analyzed by real-time PCR. (B) DC2.4 cells were infected with T. gondii or treated with TgESP for 24 hr. NOX4 levels were measured by western blot and RT-PCR. (C, D) DC2.4 cells were stimulated with GFP-RH or TgESP for 24 hr and treated with PI3K or AKT inhibitor during final 2 hr. NOX4 levels were confirmed by (C) western blot or (D) fluorescence microscopy. Scale bar=10 μm.
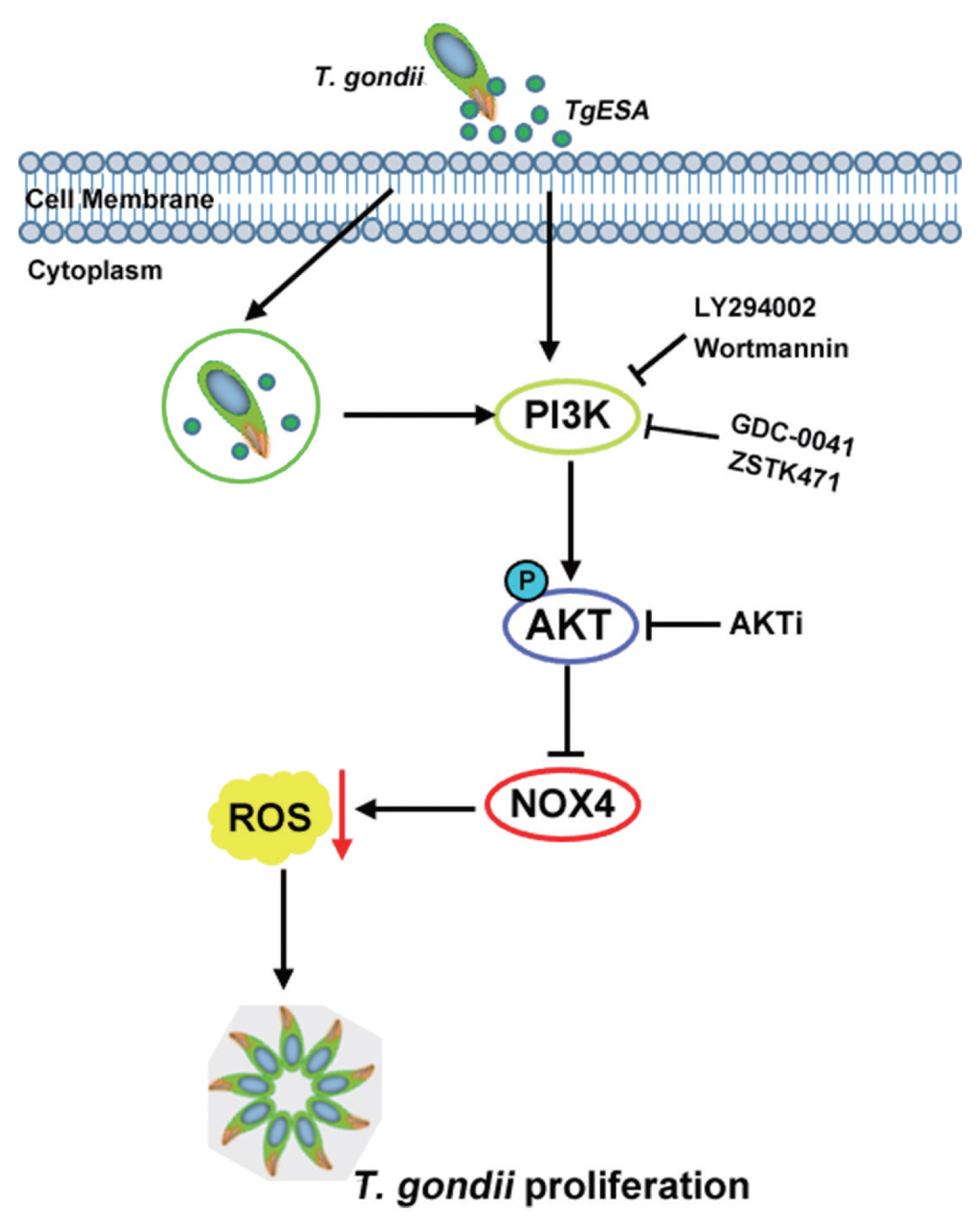
Model for dendritic cell environment modification by T. gondii.
DISCUSSION
ROS regulation in host immune cell is important for the survival and growth of T. gondii, but how T. gondii regulates host intracellular signalings and ROS level are not well-known. Therefore, we screened for a host signaling system which can be controlled by T. gondii and which can regulate the ROS level in dendritic cells.
In order to study the T. gondii induced ROS regulation mechanism in dendritic cell among various host signal pathways, PI3K/AKT pathway was selected as our target based on the reports that infection with T. gondii can inhibit host apoptosis by activating AKT in host cells [16,17,21], and in hematopoietic cells, T. gondii-activated host AKT inhibited autophagy in infected cells [15]. Previously we have reported that T. gondii infection reduce ROS in the host cells retinal pigment epithelium cell line (ARPE-19), in PI3K/AKT activity dependent manner [20].
According to the data described previously, T. gondii infection or TgESA treatment were able to induce AKT phosphorylation of DC2.4 cells (Fig. 1A), and the reduction of AKT phosphorylation by treatment with PI3K-specific inhibitors indicate that T. gondii-induced AKT phosphorylation in dendritic cells is under control of PI3K activity (Fig. 1B). Interestingly, AKT activation of DC2.4 cell by T. gondii infection not only reduced the endogenous ROS in the host cell but also was capable of suppressing the H2O2-induced ROS (Fig. 3A–E). To the contrary, inhibiting PI3K/AKT signaling pathway activity by the chemical inhibitors reversed T. gondii-induced host ROS suppression (Fig. 3C). These results indicate that PI3K/AKT pathway activity is indispensable for the reduction of ROS in DC2.4 cells by T. gondii infection.
Kim and her colleagues have reported that T. gondii infection in bone marrow-derived macrophage (BMDM) at the early period (1 hr infection) increases the amount of ROS in host cells and that increased ROS induces the expression of macrophage migration inhibitory factor (MIF), which inhibits proliferation of T. gondii [22]. The authors reported that intracellular ROS-dependent MIF expression is essential for macrophages-regulated host defenses against T. gondii infection, and that NOX4 KO mouse is more susceptible to parasite infection and growth [22]. There is a difference between Kim and our reports in the aspect of host ROS profile change by T. gondii. Our observation is made with host infected for 24 hr, unlike Kim’s observation which is done in 1 hr. This difference also suggests that unlike early infection response, long time infection (24 hr) may show inverse correlation between the number of T. gondii and host ROS profile. NOX4 is a member of NADPH oxidase family which are known to generate ROS, for example, in phagocytosis [23]. Electron microscopy measurement of cytochemical NAD(P)H-oxidase activity using CaCl2 showed that HUVEC stimulated with IFNγ increased phagocytosis and increased NADPH oxidase activity at the site and inhibited proliferation of T. gondii [24]. These reports suggest that ROS expressed by NADPH oxidase may be important for suppression of T. gondii proliferation. However, parasite needs methods to overcome the ROS attack from the host, and we presumed that manipulation of host PI3K/AKT pathway by T. gondii will be a solution. Indeed, when the AKT activity of host cells was inhibited by PI3K and AKT inhibitors, T. gondii infection or TgESA treatment had no effect on host ROS level (Fig. 3C). Interestingly, when these data were compared with the results of the T. gondii proliferation experiment (Fig. 2A), we found that inhibition of AKT activity restored the basal level of ROS in host cells despite T. gondii infection and consequently inhibited proliferation of T. gondii. The finding confirmed that the AKT activation in host cells results in ROS reduction, thus creating a favorable environment for the proliferation of T. gondii. Invasion and attachment assay, contrarily, showed no change in the T. gondii’s host invasion rate despite the inhibition of AKT activity by PI3K inhibitor (Fig. 2B). In other words, invasion or attachment of T. gondii into host cells are independent events of host PI3K/AKT activity.
Based on the result, a question was derived that what could be the specific mechanisms that can regulate host ROS, for example, the transcription factors or the ROS sources which can be controlled by them. Through the reference search, we were able to converge the candidates down to NADPH oxidases (NOXs). In human the NOX family consists of 7 members, NOX 1-5 and DUOX 1-2, but NOX 5 is not present in rodents [25]. These NOX family enzymes are known to be involved in producing ROS, which is essential in various catabolic activities including phagolysosome process. It is also reported that if a defect occurs in the subunits of NADPH oxidase subunits, variety of problems may arise in the ROS generation, resulting in chronic granulomatous disease (CGD) [23] which increases the body’s susceptibility to infections caused by certain bacteria and fungi.
We have shown that PI3K/AKT activity induced by T. gondii infection can negatively regulate NOX family expression in dendritic cells, as in epithelial cells. We previously reported the possibility using the T. gondii-RPE infection model that T. gondii can reduce host NOX4 gene expression in PI3K/AKT dependent manner [20]. Additionally, the presence of 2 potential FOXO transcription factors specific binding sequences in the murine NOX4 promoter site has been revealed through in silico analysis. These findings support the idea that T. gondii may reduce host ROS production via PI3K/AKT signaling activitation, and create a favorable environment for its growth. It is also confirmed that PI3K/AKT activity is irrelevant with the invasion activity of T. gondii to host cell, but is indispensable for T. gondii’s intracellular replication which will be reflected as parasitemia level in the host. Based on the results, we propose the possibility that PI3K/AKT inhibition is an important therapeutic target for developing anti-T. gondii drug or cure for inflammatory symptoms.
ACKNOWLEDGEMENTS
This work was supported by the Research Fund of Chungnam National University.
Notes
CONFLICT OF INTEREST
The authors declare no conflict of interest related to this study.