Comparative Assessment of Diagnostic Performance of Cytochrome Oxidase Multiplex PCR and 18S rRNA Nested PCR
Article information

Abstract
Malaria elimination and control require prompt and accurate diagnosis for treatment plan. Since microscopy and rapid diagnostic test (RDT) are not sensitive particularly for diagnosing low parasitemia, highly sensitive diagnostic tools are required for accurate treatment. Molecular diagnosis of malaria is commonly carried out by nested polymerase chain reaction (PCR) targeting 18S rRNA gene, while this technique involves long turnaround time and multiple steps leading to false positive results. To overcome these drawbacks, we compared highly sensitive cytochrome oxidase gene-based single-step multiplex reaction with 18S rRNA nested PCR. Cytochrome oxidase (cox) genes of P. falciparum (cox-III) and P. vivax (cox-I) were compared with 18S rRNA gene nested PCR and microscopy. Cox gene multiplex PCR was found to be highly specific and sensitive, enhancing the detection limit of mixed infections. Cox gene multiplex PCR showed a sensitivity of 100% and a specificity of 97%. This approach can be used as an alternative diagnostic method as it offers higher diagnostic performance and is amenable to high throughput scaling up for a larger sample size at low cost.
Malaria is one of the most devastating parasitic diseases in the world caused by the genus Plasmodium transmitted between human and a hematophagous host, Anopheles. Among the 5 species of Plasmodium, P. falciparum and P. vivax pose serious public health challenge. P. falciparum is the most virulent species responsible for the majority of malaria cases. World Health Organisation estimated approximately 5.6 million cases of malaria infection in India in the year 2019 [1].
Malaria treatment is completely based on an accurate and reliable diagnosis. Identification of correct species, diagnosis of low parasite densities, and mixed infections can contribute to accomplishing malaria elimination. The goal of malaria elimination requires new interventions that can address residual malaria transmission as well as new tools to monitor their impact on vector-borne disease transmission.
In various regions across India, majority of malaria cases is caused by P. falciparum or P. vivax infections. They are unevenly distributed throughout the country. In few endemic areas, both the species coexist [5]. This situation needs an accurate and reliable method of diagnosis. Hence, a prompt, robust, and cost-effective method is required for the detection of Plasmodium species. Microscopy is considered as a gold standard test for the malaria diagnosis [3,6]. However, species determination in low parasitemia conditions is challenging, laborious, and ill-suited for high-throughput use [4]. Rapid Diagnostic Tests (RDTs) are increasingly being used because they are accurate, sensitive, cost-effective, and easy to application. Among various other causes, genetic variability in diagnostic antigens and deletions of histidine rich protein 2 (HRP2) and HRP3 in P. falciparum population lead to false negative results [3,7]. Persistence of HRP2 in the blood, poor detection at very low parasitemia, and inability to quantify the parasites are few other drawbacks of RDTs [2].
Various molecular techniques such as polymerase chain reaction (PCR), microarray, mass-spectrometry, flow cytometry, loop-mediated isothermal amplification (LAMP) assay, and real-time PCR are available for the diagnosis of malaria [8–10]. Nested PCR targeting 18S rRNA is considered as a gold standard for malaria diagnosis, while it included several limitations, such as long turnaround time and multiple steps allowing exposure of the sample to the environment that increases the chance of false positive results. LAMP comprises a set of 6 primers for target amplification under constant temperature. However, in the case of potential contamination, there is no specific indicator to determine whether the result is true or false positive. Real-time PCR is a rapid, quantitative, and reliable test, but reagent cost and instrument cost are much higher compared to conventional PCR or LAMP, particularly when there is a large number of samples. Hence, we compared Cytochrome oxidase (cox) gene-based multiplex PCR, which can detect as low as 0.02 pg/μl compared with 18S rRNA gene species-specific nested PCR with the limit of 10 p/μl [11,12].
This study was approved by ethics committee of ICMR-NIMR, New Delhi, who waived the requirement of written informed consent (ECR/NIMR/EC/2017/89).
A total of 120 samples were collected from Odisha on Whatman filter paper during a previous cross sectional study [13]. Few drops of blood were collected by the finger prick to prepare blood smears and dried blood spots (DBS). Positive and negative controls for the experiment were obtained from the Parasite Bank of ICMR-National Institute of Malaria Research, New Delhi.
Thick and thin blood smears were stained with Giemsa solution and screened for malarial parasites by microscopy using oil immersion lens. Blood films were examined by 2 microscopists who were blinded to each other’s result.
Rapid diagnostic tests were performed in all the blood samples using bivalent malaria RDT kits Malarigen (Aspen laboratories, Delhi, India). With the help of blood transfer device supplied with the test cassettes, 5 μl blood was dispensed into the sample well and buffer was added according to the manufacturer’s instructions. The test was scored as positive if either of the P. falciparum or the P. vivax lines together with the control line were visible and negative if only the control line appeared.
Malaria genomic DNA was isolated from the blood samples using the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions and stored at −20°C. Concentration and purity of isolated genomic DNA were quantified by Nanodrop 2000 spectrometer. Standard PCR was performed targeting 18S rRNA gene using primers as previously described. [12]. A total reaction volume of 20 μl was prepared containing 1.8 mM MgCl2, 250 μM of each dNTPs, 1 μM each of forward and reverse primers, 2 μl of PCR buffer (10 mM Tris–HCl, pH 8.3, 50 mM KCl), 1.0 unit of Taq DNA polymerase, and 2 μl of genomic DNA, and the remaining volume was adjusted by nuclease free water. All amplifications were performed in a thermal cycler (ABS, Waltham, Massachusetts, USA) with following amplification conditions: denaturation cycle at 96°C for 10 min; 30 cycles at 95°C for 1 min, and 55°C for 5 min; and a final extension at 60°C for 1 h. Fragment extended by rPLU5 and rPLU6 was amplified. Next, 2 μl of the ‘nested 1’ amplified product was used to prepare dilution ratio 1:50. Twenty microliters of diluted PCR product was used as a template DNA for secondary amplification reaction (nested 2), wherein the species-specific (rVIV1–rVIV2 and rFAL1–rFAL2) primer pairs were used for each of the 2 separate reactions. Amplified products from nested 2 were analysed using electrophoresis on 1.5% agarose gel stained with ethidium bromide.
Cox gene multiplex PCR was performed to detect P. falciparum and P. vivax [11]. PCR cycler profile included denaturation at 96°C for 10 min, 30 cycles at 95°C for 1 min, and 55°C for 5 min; and a final extension at 60°C for 1 h. The reactions were based on the sequences of P. falciparum, cox III (Access no. GI8346992 and M76611) and P. vivax, cox I (Access no. GI63022502). The nucleotide sequences of cox I and III genes were used as previously mentioned [11]. Multiplex PCR using both the primers were initially optimized on positive controls of P. falciparum and P. vivax. Amplified PCR products were analysed using electrophoresis on a 2.5% agarose gel stained with ethidium bromide.
Discordant P. falciparum-positive samples detected by Cox gene multiplex PCR and 18S rRNA samples were cross validated. PfMSP2 gene was amplified for P. falciparum samples and PvCSP gene for P. vivax [14,15]. Diagnostic performance was evaluated for cox gene multiplex PCR with that obtained from standard microscopy. Statistical analysis was carried out using SAS University edition and Microsoft Excel. The Proc-Freq function was used to calculate sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), and confidence intervals. Proc-Logistic function was used to plot the receiver operating characteristic (ROC) curves. The ROC curve shows the trade-off between sensitivity (TPR) and specificity (1–FPR).
A total of 120 afebrile individual samples were diagnosed using microscopy, RDT, 18s rRNA PCR, and cox gene multiplex PCR (Table 1). Parasite identification from microscope, including P. falciparum (7.5%) and P. vivax (4.1%), was found to be 11.7%. P. falciparum density ranged between 380–490,560 parasite/μl (geometric mean: 245,470 parasite/μl) and P. vivax density ranged between 420–32,052 parasite/μl (geometric mean: 16,236 parasite/μl). In order to evaluate diagnostic performance, 12 samples from P. falciparum positive by cox gene multiplex PCR, Nested PCR, and microscopy were used. In multiplex PCR, 8 samples were positive to P. vivax and 7 samples were positive by nested PCR (Fig. 1). Microscopy and RDT detection rates were similar each other in diagnosing P. vivax. Out of 120 samples, 4 samples were found to be mixed infection of P. falciparum and P. vivax by cox multiplex PCR, but only 2 samples were found to be mixed infection by nested PCR (Table 1). Discordant results obtained between 18S rRNA nested PCR and cox gene multiplex PCR were validated by amplifying PfMSP2 and PvCSP gene, which revealed cox gene multiplex PCR results to be accurate [14,15]. The sensitivity of cox gene multiplex PCR was 66.7%. In contrast, microscopy and RDT showed 100% specificity, and 18S rRNA and cox gene multiplex PCR showed 97% specificity. PPV for 18S rRNA and cox gene multiplex PCR was 87.5%, whereas NPV was 100% (Table 2). AUC for18S rRNA nested PCR and cox gene multiplex PCR was 0.985 (Fig. 2).
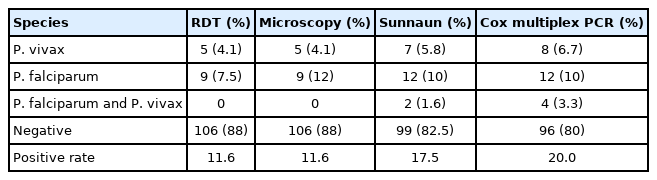
Performance of different detection methods
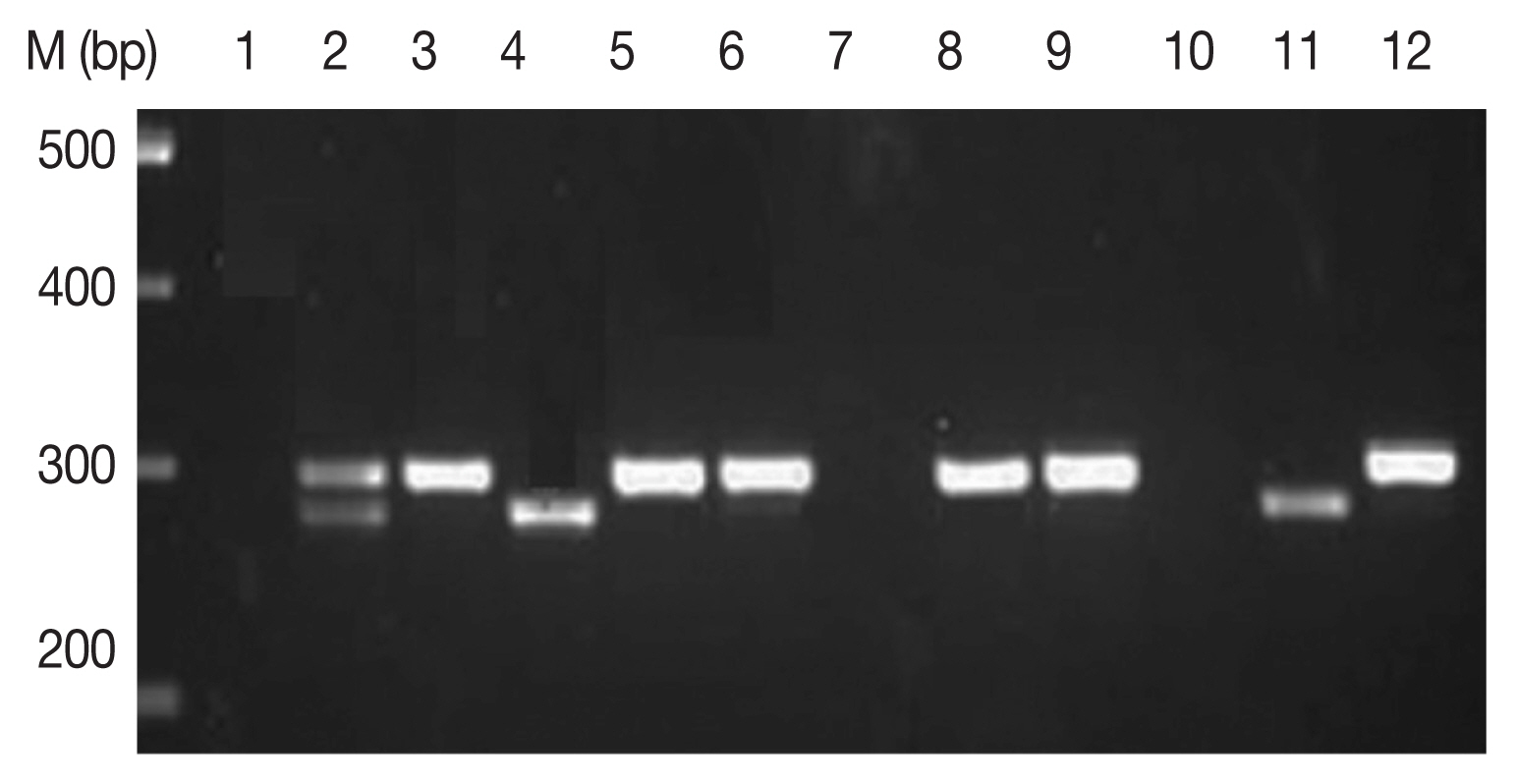
Amplification of cox 1 gene by multiplex PCR. Lane 1, P. falciparum + P. vivax (mixed) sample; lanes 2, 5, 6, 8, 9, and 12, P. falciparum (290 bp fragment); lanes 4 and 11, P. vivax amplification of 273 bp frgment.
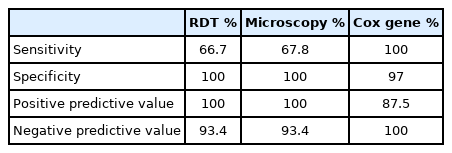
Comparison of sensitivity and specificity of malaria detection methods
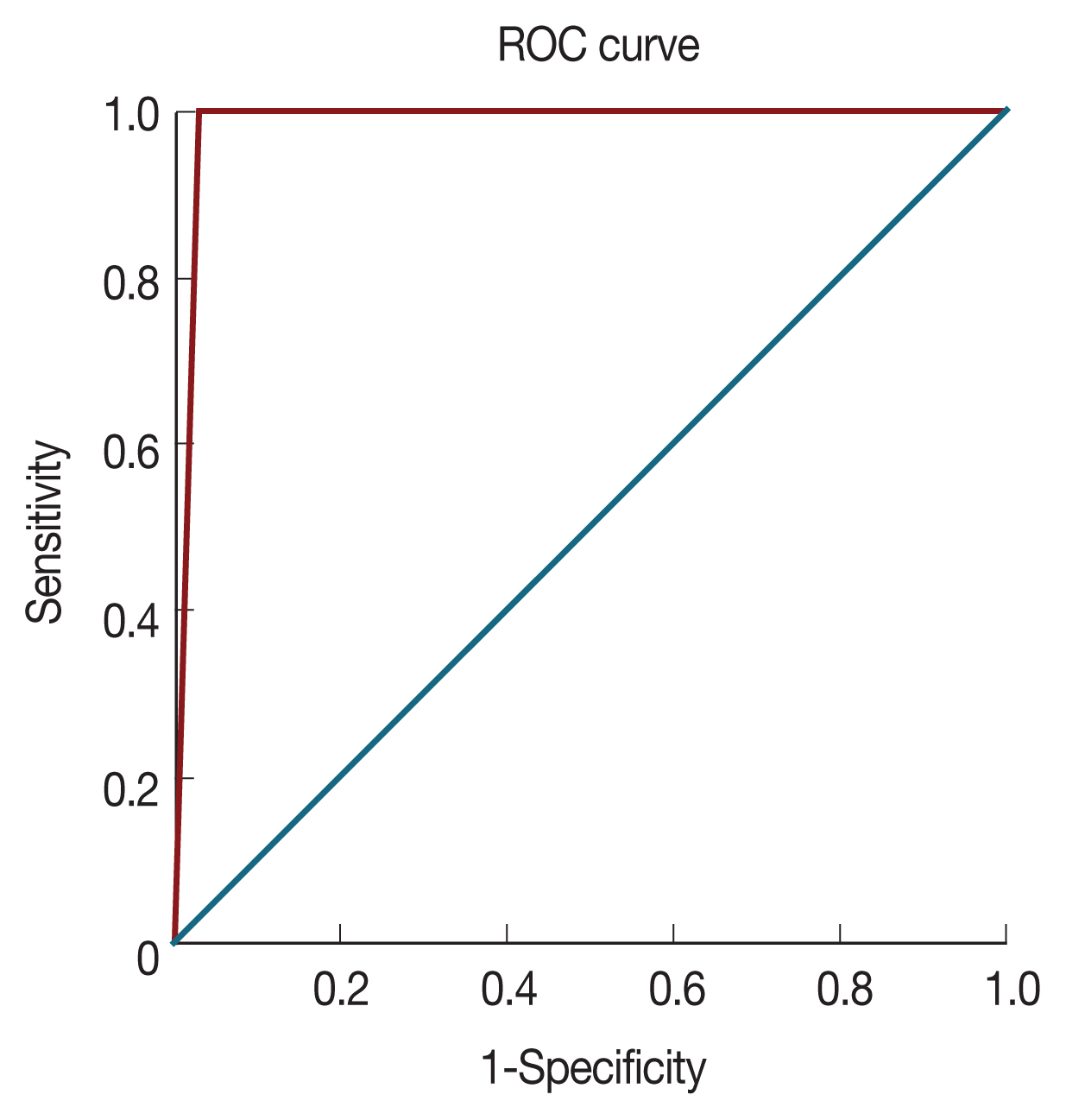
ROC curve for 18S rRNA nested PCR vs Cox multiplex PCR.
A key strategy to eliminate malaria is a prompt diagnosis, appropriate treatment, and timely follow-up. Plasmodium species identification in case of mixed infections with low parasite density is a challenge for malaria elimination. In this study, we comparatively evaluated diagnostic reliability of multiplex PCR, RDT, and microscopy. Discordant results were cross validated by PfMSP2 gene amplification and PvCSP gene. We found that cox gene multiplex PCR would represent highly true positive (Supplementary Figs. S1, 2).
High sensitivity can be achieved by targeting the genes that exist in multiple copy number in the genome [18]. Small subunit of 18S rRNA exists in 5 to 8 copies in the chromosomal genome of P. falciparum depending upon the strain. The mitochondrial DNA of P. falciparum in ring stage contains about 20 copies per cell of a 6-kb genome, while mature gametocytes have 4 to 8 copies, providing a good opportunity to detect the parasite even in low density [19].
Optimized multiplex PCR was comparatively evaluated with microscopic examination using 120 samples. We found that sensitivity of cox gene multiplex PCR was 100% (Table 2). Numerous PCR-based diagnostics have been proposed earlier, however, none showed both easy multiplex amplification and increased sensitivity compared to the standard 18S rRNA method [6,18]. Nested PCR increases specificity and sensitivity, partly because the target gene is present in a limited copy number [12]. Cox gene multiplex PCR significantly reduces cost-effectiveness, processing time, risk of contamination, and potential for technical errors compared to the nested PCR. Previous studies have reported that the sensitivity of nested PCR targeting 18S rRNA is up to 10 pg/μl of blood [12]. The samples showing discordant result between 18S RNA PCR and cox gene multiplex PCR were validated by amplifying highly conserved PfMSP2 and PvCSP genes. The sensitivity of mitochondrial PCR is 100% over nested PCR for both species [20]. Asymptomatic malaria patients or who showed no visible parasites under the microscope might have very low parasitic infections. To rule out the missed cases due to low parasite density, cox gene multiplex PCR could be a useful tool.
Multiplex PCR has great advantage while surveying cohort samples due to its high sensitivity and specificity. Compared to nested PCR, single-step cox gene multiplex PCR reduces the time factor, thereby eliminating the possibility of contamination. Cox gene multiplex PCR is an ideal, cost-effective, faster, and sensitive method for the detection of P. falciparum and P. vivax. Cox gene multiplex PCR functionally can be a better alternate diagnostic technique in research, intervention strategies, and patient monitoring. A limitation of this study was that too few positive samples. Studies with large sample sizes will help identify more sensitive and specific diagnostic modalities.
Supplementary Information
ACKNOWLEDGEMENT
Authors are thankful to the Director, ICMR-National Institute of Malaria Research, Delhi, India, for providing laboratory facilities.
Notes
All the authors declare that they have no conflict of interest.