Antimalarial activity of thiophenyl- and benzenesulfonyl-dihydroartemisinin
Article information
Abstract
Each diastereomer of 10-thiophenyl- and 10-benzenesulfonyl-dihydroartemisinin was synthesized from artemisinin in three steps, and screened against chloroquine-resistance and chloroquine-sensitive Plasmodium falciparum. Three of the four tested compounds were found to be effective. Especially, 10β-benzenesulfonyl-dihydroartemisinin showed stronger antimalarial activity than artemisinin.
The natural sesquiterpene endoperoxide artemisinin (Fig. 1-1), which was isolated from Artemisia annua L. (Klayman, 1985), has become a potential lead compound in the development of an antimalarial (Luo and Shen, 1987; Jung, 1994; Haynes and Vonwiller, 1997; Vroman et al., 1999) and recently anticancer agents (Beekman et al., 1997; Jung et al., 2003; Posner et al., 2003). The semi-synthetic, acetal-type, artemisinin derivatives (Fig. 1-3), ether and ester derivatives of trioxane lactol dihydroartemisinin (Fig. 1-2), were developed for their higher antimalarial efficacy and are now widely used to treat malarial patients (Fig. 1) (Brossi et al., 1988).
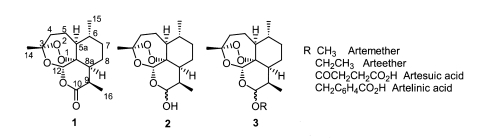
Structure of artemisinin and acetal-type artemisinin derivatives.
Although these acetal artemisinin derivatives showing potent antimalarial activity in vitro, the acetal functional group at the C-10 position is responsible for chemical instability (Jung and Lee, 1998), and toxicity (Gordi and Lepist, 2004). Therefore, to improve bioavailability, it is important to discover novel artemisinin derivatives suitable to chemo-antimalarial therapy. In 1995, Venugopalan et al. reported a series of thioacetal type artemisinin derivatives, some of which have potent antimalarial activity in vivo.
In 1998, Posner et al. also reported that sulfide and sulfone endoperoxide from R-(+)-limonene (Bachi et al., 1998) and sulfone trioxanes (Posner et al., 1998; Posner et al., 2000) have similar or less activity to natural artemisinin. However, because there is no report on C-10 sulfonyl artemisinin derivatives (Fig. 2), we decided to investigate and report the synthesis and antimalarial activity of such derivatives.

Structure of thioacetal artemisinin derivatives.
TESTED COMPOUNDS
As seen in Fig. 3, separable diastereomeric mixtures of 10α- (Fig. 3-6) and 10β-thiophenyl-dihydroartemisinins (Fig. 3-6) were prepared by reacting known dihydroartemisinin (Fig. 1-2) (Lin et al., 1987) with thiophenol (2eq) under the catalysis of BF3Et2O (1eq) at room temperature for 10 minutes (Venugopalan et al., 1995; Oh et al., 2004a, 2004b). The thioacetal products (Fig. 3-6) were transformed to produce 10α- (Fig. 3-7) and 10β-benzenesulfonyl-dihydroartemisinins (Fig. 3-8), respectively, in good yields, by performing oxidation with H2O2/urea complex (UHP), trifluoroacetic anhydride (TFAA) and NaHCO3 (Varma and Naicker, 1999; Caron et al., 2000).
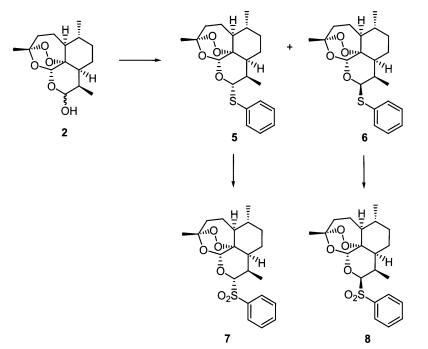
Synthesis of thioacetal artemisinin derivatives.
Strains of Plasmodium falciparum
Two culture-adapted strains of P. falciparum were used: the chloroquine-sensitive strain FCR-8/West African, and chloroquine-resistant strain FCR-3/Gambia subline F-86 of P. falciparum obtained from ATCC (Nguyen-Dinh and Trager, 1980; Jensen and Trager, 1978). The medium used was RPMI medium containing hypoxanthine and supplemented with HEPES buffer, sodium bicarbonate, human A serum, glutamine, gentamicin, and uninfected human O erythrocytes.
In vitro measurement of parasite growth inhibition by drugs
The assays were conducted in vitro by a modification of the semiautomated microdilution technique of Desjardins et al. (1979) and Delhaes et al. (2002) based on radiolabeled [3H]hypoxanthine incorporation. Drug testing was carried out in 96-well, microtiter plates. Stock solutions of each compound were prediluted in complete culture medium (RPMI 1640 supplemented with 10% pooled human A serum), and titrated in duplicate in serial twofold dilutions. The final concentrations ranged from 1.96-250 nmole L-1 for artemisinin derivatives and artemisinin, and 11.2-1435nmole L-1 for chloroquine. After the addition of a suspension of parasitized erythrocytes in complete culture medium (200µl per well, 0.7% initial parasitemia with a majority of ring stages, 1.8-2% haematocrit) and [H3]hypoxanthine (Amersham, UK, TRK74, 1µl per well), the test plates were incubated at 37℃ for 24 h in an atmosphere of 5% O2, 5% CO2, and 90% N2. Growth of the parasites was estimated from the incorporation of radiolabeled [H3]hypoxanthine into the parasites` nucleic acids, measured in a liquid scintillation spectrometer (Packard, USA). The 50% inhibitory concentration (IC50) values refer to the molar concentrations of drug causing a 50% reduction in [H3]hypoxanthine incorporation, compared to drug-free control wells. IC50 values were estimated by linear regression analysis of log-dose-response curves.
In the screening of the two standard molecules, chloroquine and artemisinin, against chloroquine-resistance (50005 = FCR-3) and -sensitive (50028 = FCR-8) parasites, we could confirm the biological property of each cell line and the inhibitory activity of each drug (Table 1). At first, two diastereomers, 10α-(Fig. 3-5) and 10β-thiophenyl-dihydroartemisinins (Fig. 3-6), had a similar inhibition activity with artemisinin. Interestingly, 10α- (Fig. 3-7) and 10β-benzenesulfonyl-dihydroartemisinins (Fig. 3-8) showed different inhibitory activity against each cell line according to the change of stereochemistry in the C-10 position of artemisinin. 10β-Diastereomer (Fig. 3-8) was ten times more active than 10α-diastereomer (Fig. 3-7). In particular, 10β-benzenesulfonyl-dihydroartemisinin (Fig. 3-8) was two times more active than artemisinin and ninety times more than chloroquine. This preliminary screening of each thiophenyl- and benzenesulfonyl-dihydroartemisinin derivative indicated that 10β-sulfonyl-dihydroartemisinin derivative can be a potentially promising antimalarial drug against chloroquine-resistance parasites.

Antimalarial activity of thiophenyl- and benzenesulfonyl-dihydroartemisinin against chloroquine-resistance (FCR-3) and -sensitive P. falciparum (FCR-8)
References
Notes
This study was supported by a grant from the Korea Science and Engineering Foundation [R05-2002-000-00808-0(2002)]