Introduction
Pakistan is a malaria-endemic country that has experienced, due to frequent floods and weak disaster preparedness, a significant rise in malaria cases in recent years. In 2022, the number of suspected malaria cases increased to >3.4 million, compared to 2.6 million estimated cases in 2021. Two Plasmodium species, P. falciparum and P. vivax, have been reported in Pakistan, with the latter predominating (>80%) [1].
The variant surface antigens (VSAs) of many Plasmodium spp. are considered the key proteins used by the parasite to escape the host immune system [2]. These proteins are encoded by multigene families located on telomeric and subtelomeric regions of the parasite,s chromosomes. Similar to the var gene superfamily in P. falciparum, the multigene superfamily of variant interspersed repeats (vir) in P. vivax encodes proteins used for avoiding the host immune system [3,4]. The vir gene superfamily consists of 346 genes divided into 12 subfamilies A–L [5]. VSAs are potential targets for vaccine development; however, the lack of an in vitro culture system for P. vivax and the variant nature of VIRs have presented challenges to their application as a vaccine or diagnostic tool [6].
Despite the variant nature of VAR proteins, vaccine trials have been partially established [7]. VIR proteins can protect against sequestration through the use of disrupting rosettes [8]. Additionally, these proteins induce moderate natural acquisition of antibody and cellular responses in P. vivax-endemic countries [9]. Like var genes, vir genes are potent candidates for vaccination against vivax malaria [5]. Since researching the genetic variation of VSAs is important, the genetic diversity of vir genes in the Republic of Korea (Korea), India, and Myanmar has been previously evaluated [10–13]. Here, we analyzed the genetic characteristics of vir genes in the P. vivax population of Pakistan and evaluated the population genetics of vir genes among different Asian populations. Our aim was to obtain in-depth information on the population structure of P. vivax, which can then be used for the development of vaccines or diagnostic tools.
Materials and Methods
Ethics statement
The Ethics Committee of Abdul Wali Khan University Mardan approved this study protocol (AWKUM/Biochem/Dept/Commit/ECR/808).
Sample collection and genomic DNA extraction
We collected a total of 89 blood samples from Khyber Pakhtunkhwa province of Pakistan between 2020 and 2021. P. vivax infection was diagnosed by rapid diagnostic test kits and microscopic examination. The patients’ blood samples were spotted on filter paper discs (Whatman 3 mm, GE Healthcare, Chicago, IL, USA) prior to drug treatment, and then they were air-dried and stored in sealed plastic bags at ambient temperature until use. Genomic DNA of P. vivax was extracted from each blood spot using a QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. The extracted DNA was preserved at −70ºC until subsequent use. Whether infections were due to only P. vivax or mixed with P. falciparum was determined by performing nested polymerase chain reaction (PCR) targeting the 18S ribosomal RNA (rRNA) gene as previously described [14].
PCR amplification and sequencing of vir genes
Four vir genes (vir 4, vir 12, vir 21, and vir 27) were selected based on subfamilies, sizes, and potent functions. These genes were amplified as previously described with minor modifications (Supplementary Table S1) [11]. Each amplicon was purified from the gel using an AccuPrep Gel Purification Kit (Bioneer, Daejeon, Korea), cloned into a pGEM-T TA cloning vector (Promega, Madison, WI, USA), and sequenced via the pUC/M13 forward and reverse primers using the Big-Dye Terminator ver. 3.1 sequencing kit (Applied Biosystems, Waltham, MA, USA) and an automatic ABI3730 DNA Analyzer sequencer (Applied Biosystems).
Diversity of vir genes and population genetic analysis
All of the vir gene sequences were aligned by the Clustal Omega multiple alignment function (https://www.ebi.ac.uk/jdispatcher/msa/clustalo). The segregating sites (S), number of haplotypes (H), haplotype diversity (Hd), average number of pairwise nucleotide differences (π), total number of segregating sites (Θw), and Tajima’s D value were evaluated using DnaSP ver. 6 [15]. When comparing between means, P-values <0.05 were considered statistically significant. Population genetic analyses, including pairwise fixation index (FST), analysis of molecular variance (AMOVA), haplotype frequencies, and nucleotide diversity based on Bei’s net distance (DA), were performed for vir genes from Pakistan, Myanmar, India, and Korea using Arlequin v.3.5.2.2. Global vir gene sequences for population genetic analyses were obtained from GenBank under the following accession numbers: Sal-1: AAKM01000104; Indian isolates: JQ733948-JQ33952; Korea isolates: KY608341-KY608363; Myanmar isolates: MN436008-436011 for vir 4; Sal-1: AAKM01000016; Indian isolates: JQ733953-JQ733971; Korea isolates: KY608364-KY08419; Myanmar isolates: MN436012-MN436028 for vir 12; Sal-1: AAKM01000003; Indian isolates: JQ733972-JQ733988; Korea isolates: KY608420-KY608471; MN436029-MN436044 for vir 21; Sal-1: AAKM01000041; Indian isolates: JQ73 3915-JQ733947; Korea isolates: KY608472-KY608510; Myanmar isolates: MN436045-MN4 36067 for vir 27 [10–13]. We analyzed the haplotype networks and generated network plots using PopArt version 1.7 [16].
Results
Genetic polymorphism of Pakistan vir genes
Among 4 vir genes amplified from P. vivax samples collected in Pakistan, vir 27 had the highest PCR-positivity rate (19.1%, 17/89), followed by vir 4 (18.0%, 16/89), vir 21 (9.0%, 8/89), and vir 12 (6.7%, 6/89) (Table 1).
Compared with the Sal-1 reference sequence, the 16 Pakistan vir 4 sequences featured 198 single nucleotide polymorphisms (SNPs), 34 singleton variable sites, and 164 parsimony-informative sites. This number of SNPs detected in the Pakistan collection of vir 4 is remarkably higher than that in Myanmar (9 SNPs), India (3 SNPs), and Korea (6 SNPs). The 198 vir 4 SNPs results from 129 non-synonymous substitutions in this gene. The vir 4 sequences from Pakistan have been deposited in GenBank under accession numbers PP5066 44–PP506659. Analysis of Pakistan vir 4 sequences showed 9 haplotypes, with an Hd value of 0.889; high values of π and Θw of 0.02905 and 0.04211, respectively; and a positive Tajima’s D value of 1.12321 (Table 1).
Analysis of 6 Pakistan vir 12 sequences identified a total of 34 SNPs, which is a number lower than those previously reported in Myanmar (168 SNPs), India (85 SNPs), and Korea (74 SNPs) (Table 1). We also observed 17 each of singleton sites with 2 variants and parsimony-informative sites with 2 variants in Pakistan vir 12. Compared to vir 12 of Sal-1, the Pakistan gene contains 2 nucleotide deletions at s sites: 706–720 (GGATCTGAAGGGGCA encoding GSEGA), 762–852 (GTAAAACCTGCACCTGCAAAACCTGTAGCG encoding VKPAPAKPVA), and 762–837 (GTAAAACCTGCACCTGCAAAACCTGTAGCGGCAAAACCTGTGGCGACAAGCCCAGCACCGTCAGAACGTGCACCT encoding VKPAPAKPVAAKPVATSPAPSERAP). The sequences of Pakistan vir 12 have been deposited in GenBank under accession numbers PP505464–PP505469. The H, Hd, π, Θw, and Tajima’s D values for Pakistan vir 12 are 3, 0.733, 0.01816, 0.01712, and 0.38820, respectively (Table 1).
Analysis of vir 21 sequences found 8 SNPs, with 5 singleton and 3 parsimony-informative sites, which are much lower than those of other regions (Table 1). Four SNPs were non-synonymous mutations. The vir 21 sequences have been deposited in GenBank under accession numbers PP505456–PP505463. The values of H, Hd, π, and Θw were 7, 0.664, 0.00244, and 0.00282, respectively, and Tajima’s D value was −0.63763 (Table 1).
Compared to the Sal-1 reference sequence, analysis of Pakistan vir 27 sequences found 25 SNPs, with 22 singleton variable and 3 parsimony-informative sites. This number of segregating sites in Pakistan vir 27 is higher than those from India (22 SNPs) and Korea (10 SNPs) but lower than that from Myanmar (32 SNPs). Thirteen Pakistan vir 27 SNPs are non-synonymous mutations. The sequences of Pakistan vir 27 have been deposited in GenBank under accession numbers PP505470–PP505486. The H, Hd, π, Θw, and Tajima’s D values for Pakistan vir 27 are 11, 0.682, 0.00278, 0.00584, and −2.10836 (P<0.05), respectively (Table 1).
Nucleotide variability of vir genes in Asian P. vivax isolates
Analyses of 48, 85, 93, and 112 sequences of respective vir 4, vir 12, vir 21, and vir 27 from 4 Asian countries (Pakistan, India, Myanmar, and Korea) in comparison to the reference sequence of Sal-1 found 18, 35, 52, and 40 haplotypes, respectively.
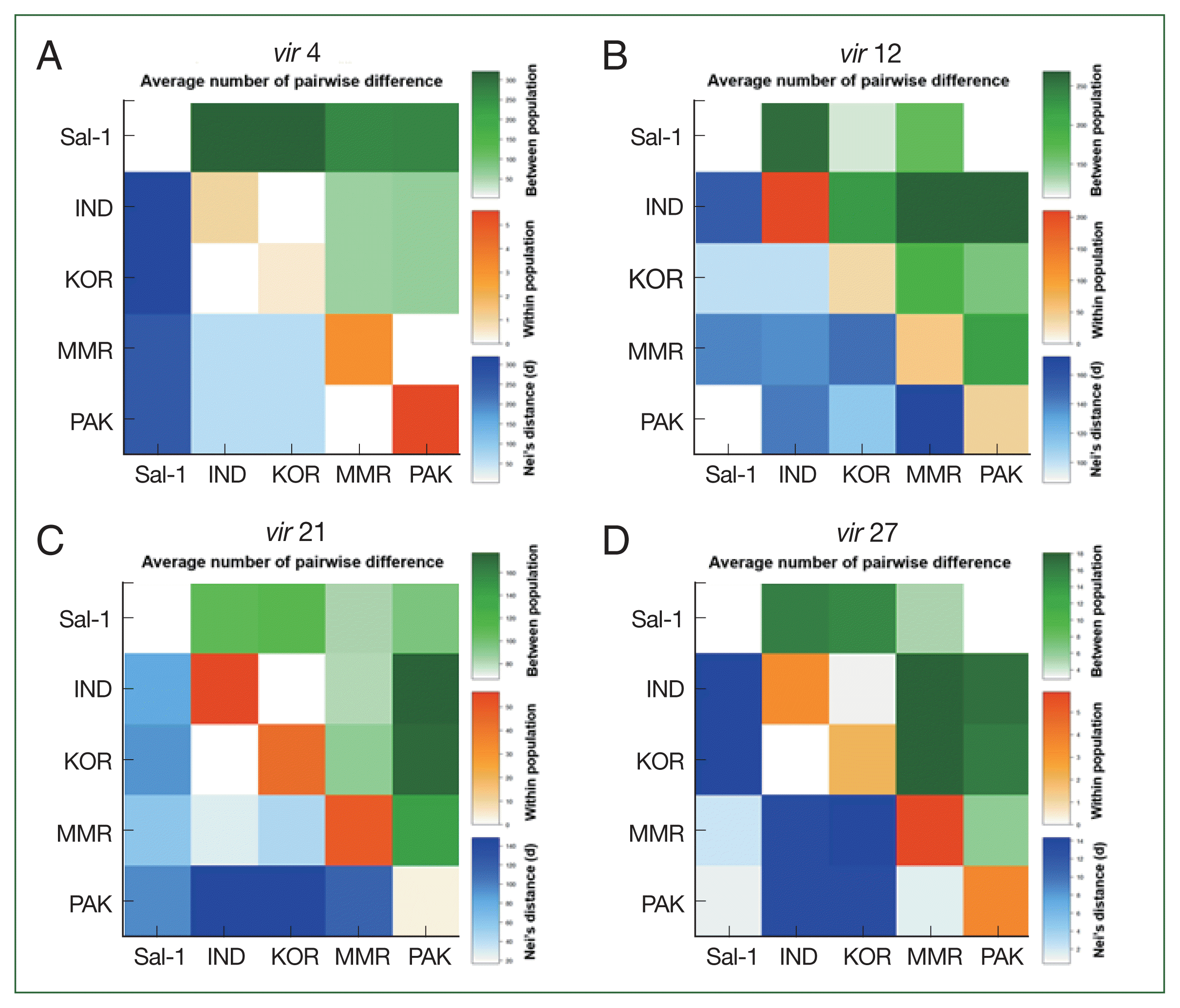
Analysis of genetic variance of vir genes among and within the Asian populations found a higher level of genetic variance among different geographic populations than within each population (Table 2). The vir 4 gene showed the highest level of variation among geographic populations (96.1%) and the lowest level of variation within populations (3.9%). In contrast, vir 21 had the lowest level of variation among geographic populations (57.1%) and the highest level of variation within populations (42.9%). Fig. 1 shows the results of pairwise difference analysis among Asian populations, with the Pakistan population of vir 4 having the higher level of genetic distinction (πxx=40.18, shown as red square in Fig. 1A) and vir 21 having the lower level of genetic distinction (πxx=3.50, shown as ivory square in Fig. 1C) compared to other populations (πxx of vir 4: 1.00, 0.51, and 3.17 for India, Korea, and Myanmar populations, respectively; and πxx of vir 21: 56.31, 43.29, and 90.28 for India, Korea, and Myanmar populations, respectively). The Indian population of vir 12 showed a greater within population genetic difference compared to those in Korea, Myanmar, and Pakistan (Fig. 1B).
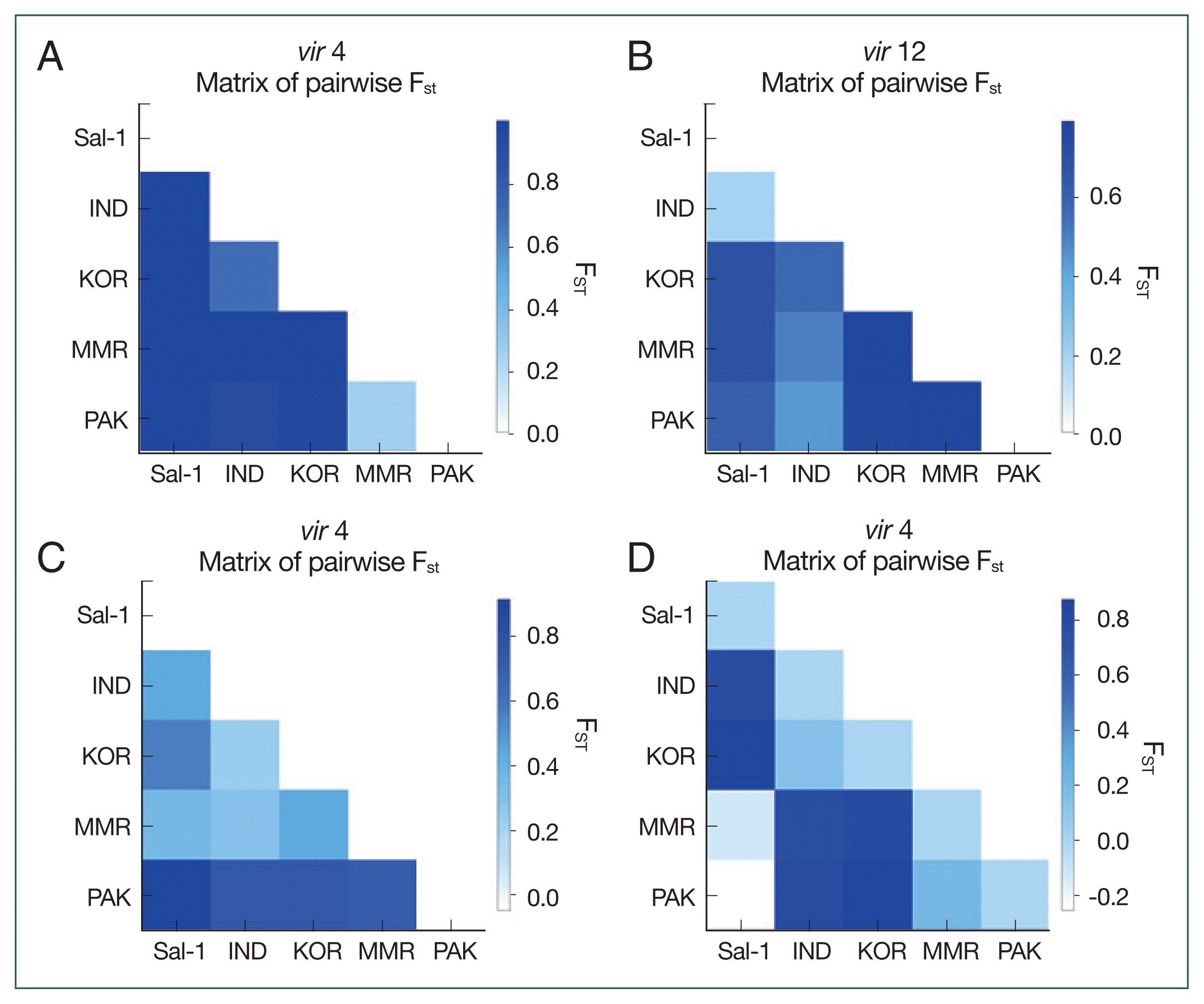
The fixation index FST was used to assess the genetic differentiation among the 4 vir genes collected from 4 different Asian populations. Fig. 2 shows different pairwise FST patterns in each vir gene. Compared to the Sal-1 reference sequence, the vir 4 gene demonstrated remarkable genetic differentiation among populations (Fig. 2A). The Pakistan population was moderately distinct from those of India and Korea; however, it was not distinct from that of Myanmar (FST=−0.01812, P-value <0.05). Compared to the Sal-1 reference sequence, the FST values for vir 12 were comparable among all populations except for that of India (FST=0.19195, P>0.05, Fig. 2B). Comparisons for vir 21 showed the Pakistan population with a higher genetic difference to the other populations, especially with the Sal-1 reference sequence (FST=0.96392, P>0.05, Fig. 2C). Comparisons for vir 27 showed the Pakistan population with a noticeable genetic distinction from those of India and Korea, but not with the Sal-1 sequence (Fig. 2D).
The haplotype network of vir genes in Asian isolates
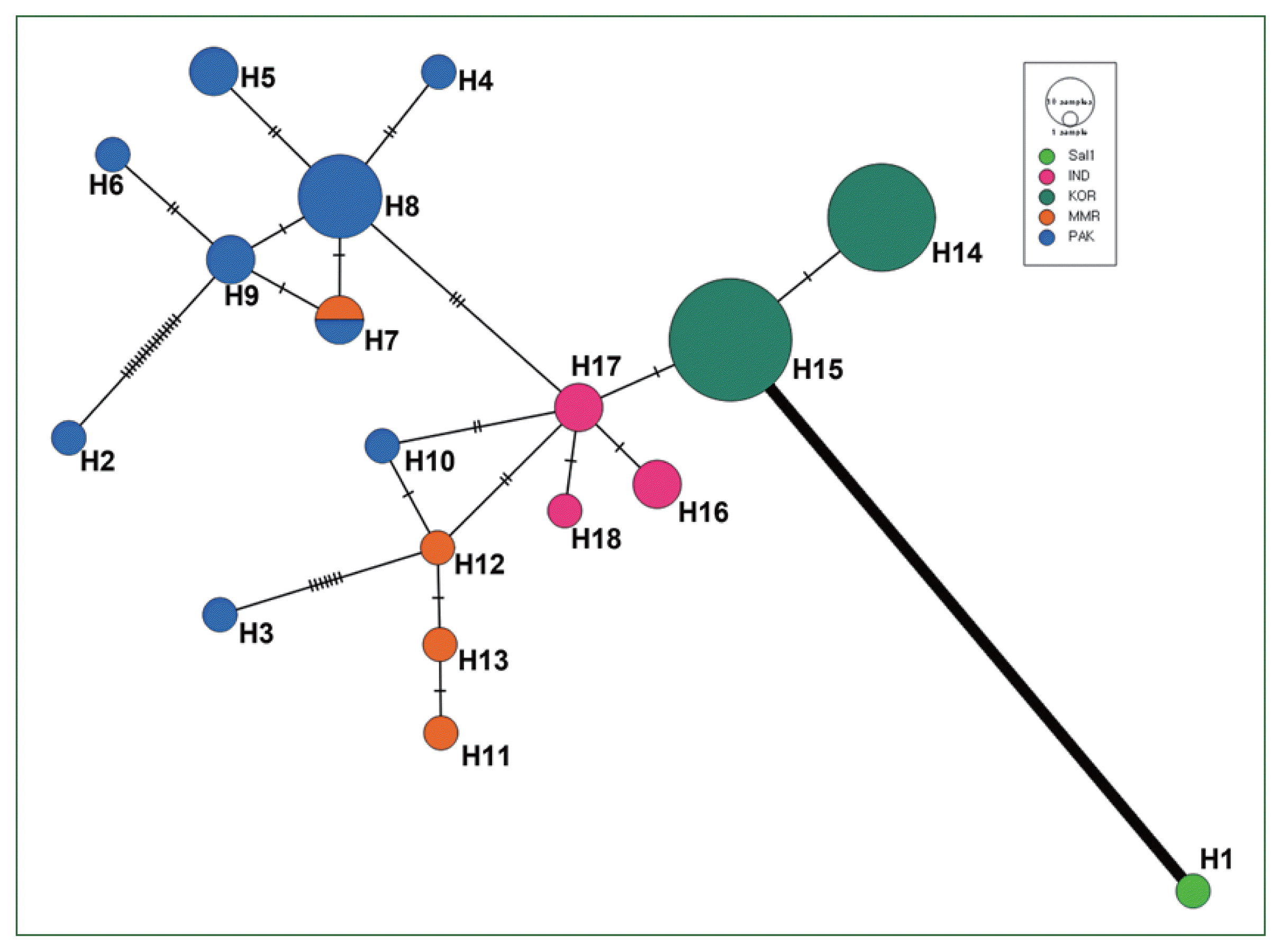
Based on the 18, 35, 52, and 40 haplotypes for vir 4, vir 12, vir 21, and vir 27, respectively, we constructed a haplotype network to characterize the frequencies and relationships of the different haplotypes. Haplotype networks, especially for vir 4, were geographically clustered (Fig. 3). All Pakistan vir 4 haplotypes were connected to the Indian and Myanmar populations, with a weaker connection to that of Korea, showing the geographical segregation of haplotypes. However, this pattern was not observed for the other vir genes. Haplotype networks of vir 12 and 21 showed that the Pakistan population was closely related to Korean and Indian populations. Meanwhile, haplotype network of vir 27 showed close relationships among all populations (Supplementary Fig. S1).
Discussion
The VIR proteins are attractive vaccine candidates because they induce a long-lasting antibody response with similar antibody levels in immune sera between singly- and multiply-infected patients without clonal expression [2]. Moreover, their cell adherence abilities suggest that they play key roles in the pathophysiology of P. vivax infection [17,18]. The genetic diversity patterns in of VIR proteins in P. vivax isolates from several countries indicate that vir genes can be used as genetic markers for the evaluation of genetic polymorphism and transmission hotspots in malaria-endemic regions in the long term [19]. Therefore, assessing the genetic diversity and population structure in global isolates is important to provide new insights into vaccine development and genetic marker potential.
Consistent with the results of previous studies [10–13], we observed different amplification positivity rates among 4 vir genes, which may be due to their location on telomeres and subtelomeres of the P. vivax genome. Genes in telomeres are very difficult to amplify because of the repetitive nature of telomeric DNA and unusual terminal structure [20]. Previous analysis of the gene diversity of the 4 vir genes have found that vir 4 is the most conserved [11,14,15]. However, our analysis of Pakistan isolates found that vir 4 is the most polymorphic gene with many segregating sites compared to the Sal-1 reference sequence. Furthermore, vir 21, which is polymorphic in other populations, was conserved among the Pakistan isolates. We attribute our findings to the dynamic molecular evolution of vir genes due to the sharp increase in malaria cases in Pakistan. This explanation is supported by the results of neutrality analyses of Pakistan P. vivax isolates, as demonstrated by the wide range of Tajima’s D values in this population (from −2.10836 for vir 27 to 1.12321 for vir 4). A positive Tajima’s D value indicates tendencies for an increase in polymorphism, a decrease in population size, and/or balancing selection. A negative Tajima’s D value tendencies for low frequency of polymorphism, increased population size, and/or purifying selection. Additionally, a Tajima’s D value below −2 or above 2 is generally a strong indication that a gene is not evolving neutrally. Genes with a Tajima’s D value below −2 have an excess of rare alleles, indicating positive selection or a selective sweep. However, genes with a Tajima’s D value above 2 have an excess of common alleles suggestive of balancing selection [21]. In contrast to vir 4, vir 27 has a Tajima’s D test value below −2, which indicates unfavorable circumstances for survival and/or in the process of being purged from the gene pool via population expansion, an effect that increases with a lower mutation rate but decreases with a higher recombination rate [22]. Furthermore, the high diversity of vir genes, along with a dramatic population expansion, indicates that the vir genes may play a role in host immune evasion, although more research is needed to explore this possibility.
Analyses of pairwise differences and the haplotype network shows that a geographical relationship among global isolates based on the vir 4 gene, and the Sal-1 reference sequence had a remarkably high pairwise difference and low genetic relationship with other Asian isolates and even haplotypes from each of the Asian countries in the network map (Fig. 3). This result is supported by the results of AMOVA analysis, which demonstrates high (96.1%) and low (3.9%) percentage variation values among and within populations, respectively, based on vir 4 characteristics. These results suggest that vir 4 can be useful as a population marker to evaluate the geographical evolution and genetic structure of P. vivax. In contrast to vir 4, we observed the formation of one node consisting of vir 21 and vir 27 haplotypes among 5 Asian countries and even Brazil (Sal-1 strain). These close relations are consistent with the relatively low pairwise difference and percentage variation values in AMOVA analysis among populations.
There is currently no useful vaccine for P. vivax malaria; however, vir genes are vaccine candidates [23]. Despite this possibility, there are only limited studies on the genetic diversity and population relationships of vir genes at the global level. Longitudinal genetic diversity studies of vir genes in P. vivax populations from diverse countries are necessary to further understand vir gene function and its application to malaria control, e.g., as vaccines.
In the present study, we investigated the genetic diversity and relationships between populations of P. vivax from 5 countries (Pakistan, Myanmar, India, Korea, and Brazil) based on analysis of 4 vir genes. Among the 4 genes, vir 4 showed remarkable genetic differentiation among populations, although its characteristics were conserved within populations, suggesting that vir 4 can be a potent marker for understanding P. vivax population structure. In contrast, vir 21 and vir 27 are characterized by a low pairwise difference and percentage variation among populations; therefore, these can be considered vaccine candidates. A better understanding of the function of vir genes and their application to malaria control, e.g., as vaccines, requires longitudinal genetic diversity studies of vir genes in samples obtained from more diverse countries.