Ticks are ectoparasites that play an important role in the transmission of various pathogens, making them significantly important in medical and veterinary sciences [1]. The accurate identification of tick species is essential for assessing the risk of tick-borne diseases because different tick species can transmit different pathogens [2]. Traditionally, the identification of tick species relies on morphological characteristics—such as body size, mouthpart shape, scutum structure, dental formula, festoon, coxa spurs, and other traits [3]. However, these morphological keys are often inadequate for identifying immature or damaged specimens [4]. Molecular techniques have emerged as valuable tools that provide objective and accurate results for species identification and evolutionary analysis.
Mitochondrial and nuclear genes are widely used for the molecular diagnosis of organisms. Mitochondrial genes are divided into protein-coding and ribosomal genes; protein-coding genes generally evolve more rapidly than ribosomal genes [5]. The cytochrome c oxidase subunit 1 (cox1) gene, a mitochondrial protein-coding gene, is considered a powerful marker for species identification and genetic variation analysis due to its rapid evolution and ease of sequence alignment [6]. The cox1 gene has proven useful in understanding tick biodiversity and tick population genetics, while the mitochondrial ribosomal gene, such as 16S rDNA, has been used to study the genetic evolution of ticks, especially among closely related species [7]. Nuclear 18S rDNA has been used to investigate evolutionary relationships among tick genera; however, due to its high degree of conservation and lack of reference databases, it is less effective in distinguishing closely related tick species [8].
To date, several studies have reported the distribution of various tick species in Korea. The most dominant tick species is Haemaphysalis longicornis, which is found nationwide, followed by H. flava and Ixodes nipponensis [9]. In addition, H. japonica and I. persulcatus have been reported in the eastern region, whereas I. granulatus and Amblyomma testudinarium have been identified in the southwestern region of the country. While many studies have relied on morphological keys for species identification [10] or on amplifying a partial fragment of the cox1 gene [11], mitochondrial 16S rDNA or nuclear 18S rDNA have remained largely unexamined in numerous cases. Moreover, in the case of cox1, only a partial fragment was analyzed, making it difficult to assess its molecular and evolutionary characteristics. Therefore, this study aimed to analyze the complete cox1, mitochondrial 16S rDNA, and nuclear 18S rDNA of ticks collected in Korea and evaluate their molecular characteristics.
Nine ticks collected in 2024 from 4 provinces in Korea (Chungcheongbuk-do, Chungcheongnam-do, Jeollabuk-do, and Jeollanam-do) were analyzed. The ticks were collected from vegetation using the flagging method and stored in 70% ethanol at room temperature until morphological identification and DNA extraction. Morphological identification was performed using a microscope according to the identification standards [3]. As shown in Table 1, the 9 ticks belonged to 3 genera and 5 species: H. longicornis (2 females), H. flava (1 male and 1 female), H. doenitzi (1 male), I. nipponensis (2 females), and A. testudinarium (2 nymphs). All ticks were unfed.
DNA was extracted from individual ticks using a DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. For molecular analysis, PCR was performed to amplify the complete cox1 gene from each tick species using newly designed primer sets (Supplementary Table S1). The complete cox1 gene was not successfully amplified by a single PCR; therefore, several short and overlapping fragments were amplified and subsequently assembled into the complete sequence. PCR was performed using the AccuPower HotStart PCR Premix Kit (Bioneer, Daejeon, Korea), and all the amplicons were sent to Macrogen (Daejeon, Korea) for direct sequencing. In addition, eukaryote-common and newly designed primer sets were used to amplify complete nuclear 18S rDNA and mitochondrial 16S rDNA, respectively, and the PCR products were subsequently cloned into Escherichia coli DH5α using the pTOP V2 vector (Enzynomics, Daejeon, Korea). At least 3 colonies per sample were selected and sequenced using a universal primer set (M13F and M13R) by Macrogen.
The obtained nucleotide sequences were aligned and assembled, and phylogenetic analyses were performed using MEGA 11 [12]. Briefly, the phylogenetic tree was constructed using the maximum-likelihood method with 500 bootstrap replications; tick sequences available in the GenBank database were included. In addition, pairwise distances were calculated using MEGA 11 with the maximum composite likelihood model. All sequences generated in the current study were submitted to the GenBank database (accession No. cox1, PV602719–PV602727; mitochondrial 16S rDNA, PV602742–PV602750; nuclear 18S rDNA, PV602732–PV602740).
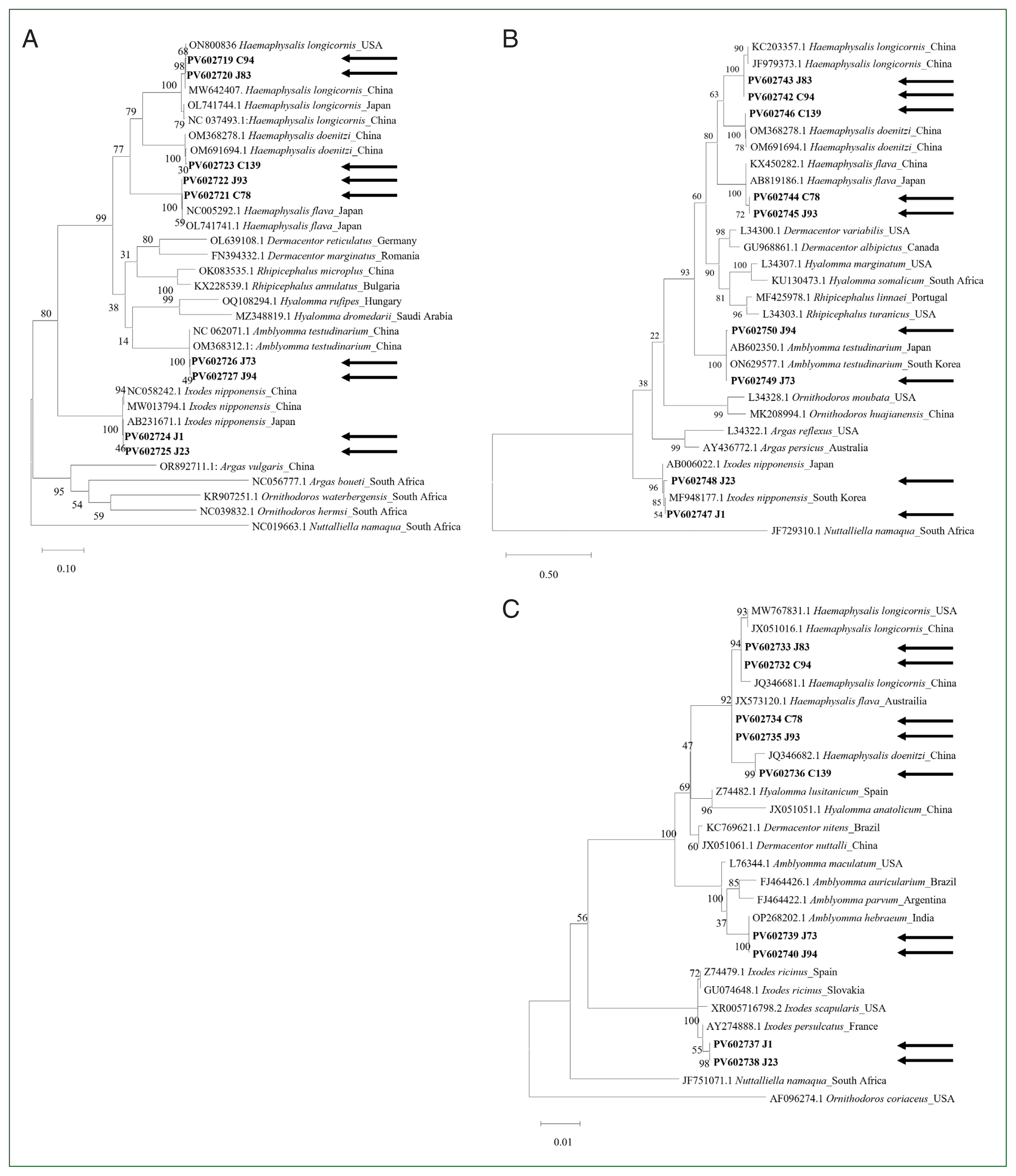
The complete cox1 gene (1,539 bp) was successfully sequenced in 5 tick species. In all tested tick species, the initial and terminal codons were consistently identified as ATT and TAA, respectively, in agreement with previous findings [13]. Notably, Haemaphysalis (30.6%–30.7%) had the highest GC content, followed by Ixodes (29.7%–29.8%) and Amblyomma (26.9%–27.0%). Pairwise distance analysis based on the cox1 gene indicated that genetic homology was high within the same genus but reduced between different genera (Supplementary Table S2). In addition, phylogenetic analysis of cox1 revealed distinct separations among tick species (Fig. 1A). The H. longicornis sequences obtained in this study were identical to those obtained from China (MW642407) and the USA (ON800836) in the GenBank database. Similarly, the H. flava sequence clustered closely with sequences from Japan (NC005292 and OL741741), and H. doenitzi clustered with sequences from China (OM691694 and OM368278). A. testudinarium was clustered with sequences from China (OM368312 and NC062071). The analysis also showed that I. nipponensis was closely related to sequences from Japan (AB231671) and China (MW013794).
The complete mitochondrial 16S rDNA gene (1,191–1,215 bp) was also successfully sequenced in this study. Pairwise distance analysis based on mitochondrial 16S rDNA revealed higher genetic homology within the same genus compared to that between different genera (Supplementary Table S3), and a phylogenetic tree showed clear species separation (Fig. 1B). The H. longicornis sequences clustered with GenBank sequences from China (JF979373 and KC203357), whereas H. flava clustered with sequences from both China (KX450282) and Japan (AB819186). Furthermore, H. doenitzi aligned with sequences from China (OM691694 and OM368278), A. testudinarium was clustered with sequences from Japan (AB602350) and Korea (ON629577), and I. nipponensis was grouped with a sequence from Korea (MF948177).
The length of the sequenced complete 18S rDNA was 1,812–1,816 bp. Unlike cox1 and mitochondrial 16S rDNA, pairwise distance analysis based on nuclear 18S rDNA revealed relatively high genetic homology, even among different genera (Supplementary Table S4). A phylogenetic analysis revealed a clear separation between tick genera (Fig. 1C). H. longicornis sequences were clustered with GenBank sequences from China (JX051016 and JQ346681) and USA (MW767831). H. flava and H. doenitzi were also clustered with sequences from Australia (JX513120) and China (JQ346682), respectively. The sequences from 2 samples, morphologically identified as A. testudinarium, clustered within the genus Amblyomma; similarly, those identified as I. nipponensis clustered within the genus Ixodes. Compared to cox1 and mitochondrial 16S rDNA, the 18S rDNA analysis was not effective in distinguishing tick species at the species level, consistent with a previous study [8].
In this study, phylogenetic analysis was performed using complete cox1, mitochondrial 16S rDNA, and nuclear 18S rDNA. Although most species were successfully identified, species identification for A. testudinarium and I. nipponensis was not possible based on 18S rDNA analysis due to the limited availability of reference sequences in databases. Nevertheless, morphological analysis, with cox1 and mitochondrial 16S rDNA analysis, confirmed that these sequences corresponded to A. testudinarium and I. nipponensis. Notably, this study is the first to report the complete nuclear 18S rDNA sequences of A. testudinarium and I. nipponensis. These results highlight the challenges often encountered in parasitological metabarcoding studies due to the limited availability of reference databases. Therefore, establishing a more comprehensive reference database is necessary to improve the reliability of molecular identification. In this context, the present study makes a fundamental contribution to improving species identification and phylogenetic analysis in tick research.
H. doenitzi is endemic to southern China [14] and distributed widely in Southeast Asia and Australia [15]. It is considered an avian tick [2] and has been reported on birds, including members of the Falconidae family in Greece [16]. In 2009, H. doenitzi was first reported in migratory birds from Hong Island in Jeonnam Province, Korea [17]. However, it has not been reported since then. The current study is the first to identify H. doenitzi on the Korean mainland using molecular information, providing valuable genetic insights into its distribution. This research contributes to a more comprehensive understanding of the molecular information and potential routes of introduction of the species, which are crucial for future epidemiological studies.
However, this study was limited by its small sample size, as only 9 ticks collected from Korea were examined. Nonetheless, the target gene is relatively well-conserved; therefore, we believe that the results of this study, including the designed primers and interspecies differences according to the genes, can be applied to the same tick species in other countries.
Analysis of the phylogenetic relationships among ticks using complete cox1, mitochondrial 16S rDNA, and nuclear 18S rDNA proved helpful in differentiating closely related hard ticks with morphological similarities. The combined analysis of cox1, 16S, and 18S rDNA sequences provides a comprehensive approach to tick identification and facilitates our ongoing efforts to enrich reference databases for improved taxonomic resolution. Additionally, this study also presents the first identification of H. doenitzi on the Korean mainland using molecular information.