Diversity of vir Genes in Plasmodium vivax from Endemic Regions in the Republic of Korea: an Initial Evaluation
Article information
Abstract
Variant surface antigens (VSAs) encoded by pir families are considered to be the key proteins used by many Plasmodium spp. to escape the host immune system by antigenic variation. This attribute of VSAs is a critical issue in the development of a novel vaccine. In this regard, a population genetic study of vir genes from Plasmodium vivax was performed in the Republic of Korea (ROK). Eighty-five venous blood samples and 4 of the vir genes, namely vir 27, vir 21, vir 12, and vir 4, were selected for study. The number of segregating sites (S), number of haplotypes (H), haplotype diversity (Hd), DNA diversity (π and Θw), and Tajima’s D test value were conducted. Phylogenetic trees of each gene were constructed. The vir 21 (S=143, H=22, Hd=0.827) was the most genetically diverse gene, and the vir 4 (S=6, H=4, Hd=0.556) was the opposite one. Tajima’s D values for vir 27 (1.08530, P>0.1), vir 12 (2.89007, P<0.01), and vir 21 (0.40782, P>0.1) were positive, and that of vir 4 (−1.32162, P>0.1) was negative. All phylogenetic trees showed 2 clades with no particular branching according to the geographical differences and cluster. This study is the first survey on the vir genes in ROK, providing information on the genetic level. The sample sequences from vir 4 showed a clear difference to the Sal-1 reference gene sequence, whereas they were very similar to those from Indian isolates.
INTRODUCTION
Plasmodium vivax is the most prevalent human malaria parasite causing most of the cases occurring outside of sub-Saharan Africa [1,2]. Vivax malaria only infects host reticulocytes, the precursor of erythrocytes, whereas Plasmodium falciparum infects all stages of erythrocytes [3]. This narrow range of infection ability of P. vivax gives it the stereotype of mild-type malaria. Until now, vivax malaria has been overlooked because of its mild characteristics compared with severe falciparum malaria [4]. However, contrary to the benign reputation of vivax malaria, its clinical manifestations are not very mild, inciting chronic infection with hypoproteinemia, edema, and coma. Furthermore, the patients with vivax malaria and concurrent nutrient deficiency can suffer from splenic rupture, which and at last, may result in death [5–7].
Since 1993, P. vivax has been consistently present in the northern areas of Gyunggido (Province) and Gangwon-do, Republic of Korea (ROK) [8,9]. Soldiers (30.5%) and veterans (25.1%) of the ROK make up over a half of the malaria patients in the country [10]. Residents (including civilians and soldiers) living near the demilitarized zone (DMZ) on the northern border of the ROK have also been infected with P. vivax. In this regard, chloroquine and primaquine have been used as a prophylactic measurement for the soldiers of the endemic area. However, such massive chemoprophylaxis using both drugs leads to a concern on an occurrence of antimalarial drug resistant P. vivax in ROK.
Ever since chloroquine-resistant P. vivax (CRPv) was first reported in Papua New Guinea [11], such strain has been reported in many other places, including India, Vietnam, and even Bolivia of South America [4,12–14], with spread occurring globally within decades. This expansion of CRPv demonstrates that once an antimalarial drug-resistant strain develops, it would soon become an international problem. Therefore, the development of malaria vaccines is receiving an attention as an alternative way to prevent vivax malaria.
By virtue of their antigenic variation, variant surface antigens (VSAs) of many Plasmodium spp. are considered to be the key proteins used to escape from the host immune system through antigenic variations [15]. These proteins are coded by multigene families located on telomeric and subtelomeric regions of the parasite’s chromosomes. The var and variant interspersed repeats (vir) gene superfamilies have been recognized in P. falciparum and P. vivax, respectively [16]. The 346 vir genes are divided into 12 subfamilies, named A to L [17,18], and are reported to be related to host immune system evasion [5]. In addition, the vir proteins were revealed to be exported to the infected erythrocyte’s surface and partially induce the adherence of P. vivax-infected reticulocytes to the endothelial cell receptor [19,20]. All together, these data suggest that the VSAs encoded by the vir genes could be candidate targets for vaccine development, but the lack of an in vitro culture system for P. vivax and the variant nature of VSAs have been an obstacle to the development of a potent vaccine [21].
Trials for developing vaccines against var-encoded proteins have been partially established, dismissing the skepticism over caused by the variance of var proteins [22]. By disrupting the erythrocyte rosettes, protection against sequestration could be established [23]. Like the var genes, vir genes are potential targets for vaccination against vivax malaria [17]. Although researching the genetic variation of VSAs is critical in the development of a novel vaccine, there has been no investigation on the subject until now in the ROK. Previously, Gupta et al. [7,24, 25] investigated the diversity of P. vivax populations from various regions in India with vir genes. Here, we performed a population genetic study in ROK. Among vir genes, the vir 27, vir 21, vir 12, and vir 4 sequences on the subtelomeric area were more specifically analyzed. This study will provide a better understanding of the diversity of vir genes.
MATERIALS AND METHODS
Ethics
The study was approved by the Ethics Committee of the Kyungpook National University. An approval form was used to obtain written informed consent from each participant. Each participant also consented to provide a 5-ml blood sample.
Sample collection
A total 85 venous blood samples were collected in the northern part of ROK, which are already known as malarial endemic regions [9], from 2011 to 2013. The samples included 5 from Gimpo (GP), 2 from Paju (PJ), and 15 from Cheorwon (CW) in 2011; 6 from GP, 23 from PJ, and 7 from CW in 2012; and 10 from PJ and 17 from CW in 2013 (Fig. 1). The collection criterion was the patient living in the study area who showed tertian fever. The blood samples were kept in EDTA-treated tube and cooled in −70°C.
Blood collection regions. From left to right, Gimpo (GP), Paju (PJ), and Choerwon (CW) are denoted by asterisks.
Diagnosis of vivax malaria
Rapid diagnostic test kits (RDTs) (NanoSign Malaria P.f/P.v from Bioland, Cheonan, Korea, and SD Bioline Ag P.f/pan Rapid from SD, Hwaseong-si, Korea) were used for the first screening. Subsequently, microscopic examinations and nested PCR were performed to confirm the tentative diagnosis of vivax malaria. The microscopic tests were performed with a thick and thin blood films stained with Giemsa, and nested PCR targeting 18S rRNA was performed as previously described [26].
DNA extraction
Genomic DNA of P. vivax was extracted from 200 μl of each blood sample using a QIAamp DNA Mini Kit (Qiagen, Valencia, California, USA) following the manufacturer’s instructions. The extracted DNA was preserved in −70°C until required.
PCR amplification of vir genes
Four of the vir genes, named vir 27, vir 21, vir 12, and vir 4, were selected (Table 1). Compared with vir 27, which belongs to subfamily I, vir 4 of subfamily C and vir 21 of subfamily B have variants with more polymorphic and distributed conserved blocks within the sequences, whereas vir 12 of subfamily E is considered to be more conserved but shown high diversity among previous studies conducted on Indian isolates [24,25]. These P. vivax sequences were amplified according to the following protocol. Each vir gene was amplified by primary and nested PCR with 4 primers for vir 27, vir 21, and vir 4, and 3 primers for vir 12, respectively. The primary PCR mixture contained 2.5 μl of 10×buffer, 2 μl of dNTPs, 1 μl of forward primer, 1 μl of reverse primer, 0.25 μl of Ex taq polymerase (Takara, Shiga, Japan), 1 μl of DNA template, and distilled water to make up the volume to 25 μl. The semi-nested PCR was performed with a similar ratio of components but with 2 μl of DNA template. The primary PCR cycle was 95°C for 5 min; 35 cycles of 95°C for 30 sec, 55°C for 30 sec, and 72°C for 1 min; and a final extension of 72°C for 7 min. The semi-nested PCR had 30 cycles of the annealing step. Electrophoresis was performed on a 1.5% agarose gel with 50 mg/ml ethidium bromide. Ladders used were 100 bp for vir 27, vir 4, and vir 21, and 1 kb for vir 12.

Information of PCR ingredients used in this study
Purification of PCR products and sequencing of vir genes
Amplicons were purified with an AccuPrep® Gel Purification Kit (Bioneer, Daejeon, Korea) for cloning, according to the manufacturer’s instructions. After gel purification, the PCR products were ligated into the pGEM-T TA cloning vector (Promega, Madison, Wisconsin, USA) and transformed into Escherichia coli DH5α competent cells. Five colonies with the recombinant plasmids per each vir gene were selected, and then, sequenced with the pUC/M13 forward and reverse primers by using a Big-Dye Terminator ver. 3.1 sequencing kit (Applied Biosystems, Foster City, California, USA) with an automatic ABI3730 DNA Analyzer sequencer (Applied Biosystems).
Sequence analysis
Acquired sequences were aligned by the Clustal Omega multiple alignment function of BioEdit [27]. The segregating sites (S), the number of haplotypes (H), haplotype diversity (Hd), the average number of pairwise nucleotide differences (π), and the total number of segregating sites (Θw) were evaluated by using DnaSP ver. 5.10.01 [28].
The π and the Θw values were used to estimate the genetic diversity. The π value indicates the randomness of the sample, whereas the Θw value indicates the equilibrium of gene segregating sites in a population [29,30]. These 2 measures are also compared as a test of neutrality. Tajima’s D test of neutrality was done with the sliding window option (window length, 100 bp; step size, 25 bp) for distinguishing whether the P. vivax DNA evolved randomly, neutrally, or not, including balancing selection or directional selection [31]. This test was calculated with DnaSP ver. 5.10.01. Any differences in the mean values were considered to be statistically significant when at P<0.05.
Phylogenetic analysis
The vir genes of P. vivax Sal-1 (GenBank accession nos. AAKM01000041.2 for vir 27, AAKM01000104.1 for vir 4, AAKM01000016.1 for vir 12, and AAKM01000003.1 for vir 21) were used as the reference. Using the minimum-evolution method and maximum composite likelihood model option, the ROK vir gene sequences were compared with the Sal-1 references, and phylogenetic trees were constructed using MEGA 7.0 [32]. Each node in the trees was supported by bootstrap analysis with 1,000 repetitions. The subjects of the phylogenetic trees are labeled with the name of the gene, sample number, collected year, and region.
RESULTS
Nested-PCR results
The nested-PCR results were evaluated according to the year and region collected as shown in Table 2. The vir 27 gene revealed the highest PCR-positive rate (72.9%), followed by vir 12 (65.7%), vir 21 (46.7%), and vir 4 (31.3%).

PCR results of venous blood samples collected from malaria patients according to year of collection and region
Specifically, vir 27 was amplified in 59.1% (13/22), 80.6% (29/36), and 74.1% (20/27) of the samples collected in 2011, 2012, and 2013, respectively. According to the sample collection year, samples from 2012 showed the highest PCR-positive rate (80.6%). Per classification by region, the GP samples had the highest rate of PCR-positive results (81.8%).
For vir 21, 43 samples that were composed of 50.0% (11/22), 47.2% (17/36), and 55.6% (15/27) of the samples collected in 2011, 2012, and 2013, respectively, were amplified indicating that these samples were PCR positive. Gene was most prevalent in the samples of 2011 and 2013. Samples from PJ and CW had the highest PCR-positive rates (54.3% and 51.3%, respectively).
For vir 12, 50.0% (11/22), 83.3% (30/36), and 66.7% (18/27) of the samples collected in 2011, 2012, and 2013, respectively, were PCR positive, showing 2012 to have the highest prevalence for this gene. Samples from PJ showed the highest PCR-positive rate at 74.3%.
For vir 4, 22.7% (5/22), 36.1% (13/36), and 29.6% (8/27) of the samples collected in 2011, 2012, and 2013, respectively, were PCR positive, again with 2012 having the highest rate for this gene. The PCR-positive rate of the GP samples was higher than that of PJ and CW (36.4%, 25.7%, and 33.3%, respectively). Gene vir 4 showed the lowest positive rate on PCR.
Variable surface protein 27 gene
Compared with the Sal-1 reference sequence, 62 vir 27 genes showed 10 S, 2 of them being singleton variable sites and 8 of them being parsimony-informative sites. The nucleotide variations T93C, G101A, C228T, T256C, A257G, A259G, G262A, A590C, T741C, and A753G were separately found (Fig. 2A). The sequences of vir 27 genes are available in GenBank (nos. KY608472-608521). The H value for vir 27 was 6, and the Hd value was 0.774.

Sequence polymorphism of vir genes in the ROK P. vivax isolates. The polymorphic nucleotides were compared to that of Sal-1 (GenBank no.: AAKM01000041.2). (A) Sequences of vir 27 gene of isolates. (B) Sequences of vir 21 gene of ROK isolates compared with 5 Indian isolates. (C) Sequences of vir 12 gene of isolates. (D) Sequences of vir 4 gene of ROK and Indian isolates. The listed sequences are indicated by dots for identical nucleotides with sequences of Sal-1, and hyphens for gaps. The locations of polymorphic sites are represented above and the nucleotide changes are marked with colors.
Nucleotide diversity values of vir 27 were 0.00238 and 0.00171 for π and Θw, respectively (Table 3; Supplement Fig. 1). Tajima’s D value of vir 27 was positive (1.08530, P>0.1; Table 3; Supplement Fig. 2).
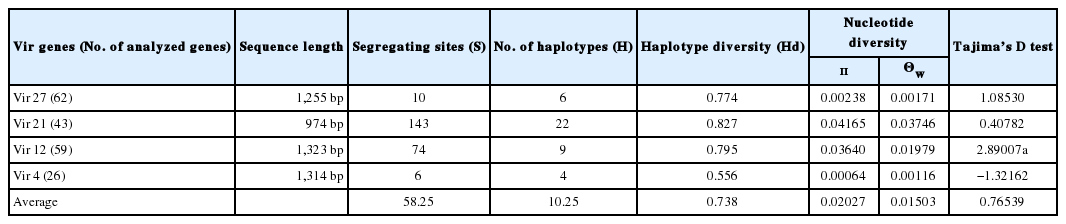
Summary statistics for the 4 vir genes in gene level
The vir 27 gene from the 62 ROK samples was divided into 2 distinct clades in phylogenetic analysis. One clade was comprised of 45 samples, including Sal-1 reference gene, and was divided into several subclades which consisted of 18 samples. In the clade of 45 samples, the main subclade was composed of 21 samples, and the other subclade was further composed of 2 clades with 10 samples each and 1 clade with 4 samples (Supplement Fig. 3A).
Variable surface protein 21 gene
There were 43 samples PCR positive for vir 21 genes. A total of 143 S were found, including 55 singleton sites with 2 variants, only 1 singleton sites with 3 variants, 84 parsimony-informative sites with 2 variants, and 3 parsimony-informative sites with 3 variants (Fig. 2B). The sequences of vir 21 genes are available in GenBank (nos. KY608420-608471). The H and Hd values were 22 and 0.827, respectively. Nucleotide diversity values were high for vir 21 (π=0.04165, Θw=0.03746) compared to those of other vir genes (Table 3; Supplement Fig. 1). Tajima’s D values for vir 21 were positive (0.40782, P>0.1; Tables 3; Supplement Fig. 2).
In the phylogenetic analysis of vir 21, an individual sample collected from CW (v21-11.27) was found. This sample was subjected to BLAST analysis whereupon it showed 99% (952/961) identity with P. vivax clone R2_21 (GenBank no. JQ733984.1) from an Indian isolate [24]. Otherwise, the difference between v21-11.27 and the Sal-1 reference gene vir 21 was 92% (882/961). With 5 Indian samples R2, R7, G9, G10, and J3 (GeneBank nos. JQ733984.1, JQ733986.1, JQ733975.1, JQ733976.1, and JQ733981.1, respectively), 2 clades were revealed on phylogenetic tree with almost the same sample sizes of 19 and 20, respectively. Compared with the clade of 19 samples, 20-sample clade had several more subclades (Supplement Fig. 3B).
Variable surface protein 12 gene
Among the 59 samples PCR positive for vir 12, there were 74 S. Six were singleton sites with 2 variants, and 68 were parsimony-informative with 2 variants, and 4 were parsimony-informative sites with 3 variants. Distinct nucleotide insertion was found at site 291, CGCTACAGGTTTTGC, and a number of nucleotides changes were also found (Fig. 2C). The sequences of vir 12 genes are available in GenBank (nos. KY608364-608419). The values of H and Hd were 9 and 0.795, respectively. Nucleotide diversity values (π and Θw) of vir 12 were 0.03640 and 0.01979, respectively. Tajima’s D value for vir 12 was 2.89007 (P<0.01; Table 3; Supplement Fig. 2).
The phylogenetic tree of vir 12 consisted of 2 separate clades. The main branch contained 3 sub-branches of 2, 15, and 17 samples, respectively. The Sal-1 reference strain was located in a second branch of 25 samples, which was divided into 2 sub-branches: 1 with 12 samples (separated with Sal-1) and the other with 12 samples (Supplement Fig. 3C).
Variable surface protein 4 gene
In the case of the 26 vir 4 gene sequences studied, 6 were S. Among nucleotide sequences, changes were found (A278C in 10 isolates, G512A in 1 isolate, and C40T, T456C, A645C, and T742A in all the isolates, respectively) (Fig. 2D). The sequences of vir 4 genes are available in GenBank (nos. KY608341-608363). The H and Hd values were 4 and 0.556, respectively. In contrast to other vir genes, the π values of vir 4 were lower than the Θw values (π=0.00064, Θw=0.00116). Moreover, vir 4 had the only negative Tajima’s D value region (Table 3; Supplement Fig. 2).
There was a distinct difference between the vir 4 genes of the 26 ROK samples and that of the Sal-1 reference strain. The ROK clade had 2 groups consisting of 16 samples and 10 samples, respectively. BLAST alignment of the ROK vir 4 genes resulted in 93–97% identity with P. vivax clone M40_4 (GenBank no.: JQ733948.1), D5 (JQ733949.1), G1 (JQ7339550.1), G2 (JQ733951.1), and G4 (JQ733952.1) from Indian isolate. With the sequences of Indian isolates, there were common mutation with Korean isolates in T742A with M40, D5, and G1. However, D5 isolate have the only mutation in G871A [24]. Otherwise, the differences between the ROK and Sal-1 vir 4 genes were 73–79% (Fig. 3).

The phylogenetic tree of vir 4 was inferred using the minimum evolution method. The optimal tree with the sum of branch length=0.00535425 is shown.
DNA diversity in regions
The π and Θw values were calculated separately for each vir gene and region (Table 4). The average π and Θw values of all vir genes were 0.02027 and 0.01503, respectively. The π values were generally higher than the Θw values except for vir 4 (π= 0.00064, Θw=0.00116). In the regional analysis, similar to the average values, the π values were higher than the Θw values in PJ and CW, but reverse was true for the GP samples.

Summary statistics for 4 vir genes with population
DISCUSSION
Plasmodium spp. have several mechanisms for host immune evasion [33]. One of them is the pir multigene superfmily, such as kir, bir, cir, and yir families encoded by Plasmodium knowlesi, Plasmodium berghei, Plasmodium chaubadi, and Plasmodium yoelii [24,34–36]. Especially in P. falciparum, these genes already studied that encoded several functional proteins known as stevor/rif and var in P. falciparum [24]. This attribute of avoiding host’s immune system can affect the severity of malaria infection in falciparum malaria by delay in immune acquisition, cytoadherence, and rosetting phenomenon [37]. A recent study showed that several vir genes have some relation with pathogenesis of vivax malaria [7]. Furthermore, cir of P. chaubadi have functional role in blood stage malaria [35].
In the present study, analysis of the vir 27 sequences of all the samples revealed that this gene is more conserved than vir 21 and vir 12, having the same nucleotide sequences as vir 27 of Sal-1 except for the mutations of 10 nucleotides. Gene vir 21 was the most polymorphic, as proven in the previous research [17]. In an earlier research, vir 12 was described as being the most conserved gene [17]. However, in our ROK samples, vir 12 was the second most polymorphic gene with many segregating sites, and high haplotype and nucleotide diversity values. All sequences of vir 4 (a member of vir subfamily C that is considered to be the most diverse) differed from vir 4 of Sal-1 at 6 mutation points. In spite of the reputation of vir 4 as the more polymorphic gene than others, this sequences from ROK seemed to be well conserved. It might be already mutated before introduction in the ROK.
The number of segregating sites ranged from as low as 6 (vir 4) to as high as 143 (vir 21). The segregating sites induced variant changes into amino acid sequences of the samples. Such changes in amino acid sequences might have affected the antigenicity of P. vivax, evading the immune system of the host. However, it is not so clear that how vir gene superfamilies’ really work on the protozoal dodge against the host attack and result in the clinical manifestation of vivax malaria [7].
Diversity parameters were similar to the number of segregating sites. First, the average number of H of all 4 vir genes was 10.25, and the average Hd value was 0.738. Gene vir 21 (H=22, Hd=0.827) was the most genetically diverse, followed by vir 12 (H=9, Hd=0.795), vir 27 (H=6, Hd=0.774), and vir 4 (H=6, Hd=0.556,). The H and Hd values of vir 4 were comparatively lower than the others, alike to a previous study in India [25]. Second, the π and Θw values of vir genes were variable from high to low. It might indicate that there was ongoing molecular mutation among vir genes of the ROK malaria isolates.
Tajima’s D test was computed for analyzing the natural selection occurrences of the P. vivax vir genes from the ROK samples. A positive Tajima D value indicates that an increase of polymorphism, a decrease of population size, and/or balancing selection can occur. A negative D value means a low frequency of polymorphism, an expansion of population size, and/or purifying selection. The vir 4 gene (−1.32162, P>0.1) had the only negative value, indicating that this gene might be under unfavorable circumstance for survival and/or in the process of being purged from the genetic pool via population expansion, an effect that increases with a lower mutation rate but decreases with a higher recombination rate [38]. With this result and the vir 4 gene’s low PCR-positive rate, the vir 4 may be disprefered to P. vivax existing in the endemic region of ROK. Tajima’s D values for vir 27 (1.08530, P>0.1), vir 12 (2.89007, P<0.01), and vir 21 (0.40782, P>0.1) were all positive, meaning a population contraction of these genes and a process of balancing selection [39]. Moreover, Tajima’s D value for vir 12 was statistically significant, indicating that among 4 vir genes this gene is under balancing selection and/or decreasing population size. In addition, such value means that vir 12 may be the major target of the host immune system in ROK. In the regional analysis, only vir 27 samples from GP had negative Tajima’s D values differ from the Tajima’s D test result of whole samples (−0.92403). In this case, vir 27 samples from GP were considered as under purifying selection, or expanding population size situation. However, it is hard to make a conclusion, because there were only 11 case collected. So, further studies will be needed. With the result of the vir 21 and vir 4 genes, there were some differences between few ROK isolates and Sal-1 vir genes. Also, those isolates were very similar to the vir genes of Indian isolates. This inferred that P. vivax malaria in the ROK might have some relationship with P. vivax in India. However, it is hard to clarifying this relationship due to lack of knowledge about vir gene superfamily.
In the current study, all phylogenetic trees showed clear 2 clades same as previous studies [24,25]. However, because of the randomly scattered sample distribution, there was no particular branching according to the geographical differences and hard to find obvious cluster.
In conclusion, this study was the first survey about the vir genes in the ROK, providing information on the genetic level. Among the 4 vir genes studied, the vir 27 was relatively conserved and expressed widely. Next, the most divergent was vir 12 whereas the most conserved was vir 4. However, the ROK sample sequences of some vir 21 and vir 4 showed a clear difference with the Sal-1 reference gene sequence, but were very similar to the vir 21 and vir 4 genes of Indian isolates. Moreover, several changes on the amino acid level were revealed. Nevertheless, it was hard to find some trend among the samples according to the period, because the study was conducted only for 3 years. Also, the revelation of the specific function of vir gene is not yet sufficient; therefore, applying VSAs from vir genes in practical areas is only at the beginning stages, and VSA variances could be an obstacle to achieving a certain product. With these results, however, the information boundary surrounding vir genes in the ROK was expanded, thereby advancing the first steps of the buildup the information basis. So, this kind of study about vir genes is needed in order to understand vivax malaria, the largest parasitic foe of healthcare.
Supplementary materials
Sliding window plot of nucleotide diversity per site (π) at vir genes (vir 27, vir 21, vir 12, and vir 4). The π values were calculated on DnaSP with window length 100 bp and step size of 25 bp.
Sliding window plot of Tajima’s D using a window size of 100 nucleotides and including each 62, 43, 59, and 26 alleles of vir 27, vir 21, vir 12, and vir 4. Positive values generally indicate that balancing selection is acting to maintain alleles at intermediate frequencies.
The phylogenetic trees were conducted by minimum evolution method. The sum of branch lengths for each optimal tree were (A) vir 27=0.01169659. The phylogenetic trees were conducted by minimum evolution method. The sum of branch lengths for each optimal tree were (B) vir 21=0.17133662. The phylogenetic trees were conducted by Minimum Evolution method. The sum of branch lengths for each optimal tree were (C) vir 12=0.09495926.
ACKNOWLEDGMENTS
This work was supported by Biomedical Research Institute grant, Kyungpook National University Hospital (2014). We are grateful to all blood donors and the staff of the Public Health Centers and hospitals in the ROK.
Notes
CONFLICT OF INTEREST
The authors declare that they have no competing interests.