Abstract
Species identification using DNA sequences is the basis for DNA taxonomy. In this study, we sequenced the ribosomal large-subunit RNA gene sequences (3,037-3,061 bp) in length of 13 Chinese Theileria stocks that were infective to cattle and sheep. The complete 28S rRNA gene is relatively difficult to amplify and its conserved region is not important for phylogenetic study. Therefore, we selected the D2-D3 region from the complete 28S rRNA sequences for phylogenetic analysis. Our analyses of 28S rRNA gene sequences showed that the 28S rRNA was useful as a phylogenetic marker for analyzing the relationships among Theileria spp. in ruminants. In addition, the D2-D3 region was a short segment that could be used instead of the whole 28S rRNA sequence during the phylogenetic analysis of Theileria, and it may be an ideal DNA barcode.
-
Key words: Theileria sp., 28S rRNA, phylogeny, cattle, sheep, China
INTRODUCTION
Members of the genus
Theileria are hemoprotozoan parasites, which have global economic and veterinary importance in ruminants. The clinical signs range from a life-threatening disease to mild or subclinical infections, depending on the infectious agent. These parasites are transmitted by hard ticks and they have complex life cycles in vertebrate and invertebrate hosts. Unlike
Babesia species, which infect only erythrocytes,
Theileria species can infect leukocytes and erythrocytes [
1]. At least 9 species of
Theileria have been reported in China. The causative agents of bovine theileriosis in China are
T. annulata,
T. orientalis,
T. mutans, and
T. sinensis [
2,
3]. Ovine theileriosis is caused mainly by
T. lestoquardi,
T. luwenshuni,
T. uilenbergi,
T. ovis, and
T. seperate [
4]. Of the ovine
Theileria present in China,
T. luwenshuni and
T. uilenbergi are considered to be the most pathogenic forms [
5,
6]. Of the bovine
Theileria spp.,
T. annulata is the most virulent species and it affects large numbers of cattle [
3,
7]. The nomenclature of the members of the benign bovine
Theileria spp. group is controversial [
8-
11]. No consensus has been reached but we used the nomenclature
T. orientalis instead of
T. sergenti/
buffeli/
orientalis in this paper [
12].
In general, the classification of members of the genus
Theileria is based on the parasite morphology, host, disease pathology, vector ticks, and geographic origin. Recently, molecular markers, such as the major piroplasma surface protein (MPSP), small subunit ribosomal RNA gene (18S), and rRNA internal transcribed spacer region (ITS), have been used in the phylogenetic analysis of
Theileria spp. [
13,
14]. The 28S rRNA forms part of the rRNA transcriptional unit, which occurs in tandem repeats arranged in ribosomal clusters throughout the genome [
15]. Recently, several molecular phylogeny studies have focused on 28S rRNA [
16-
18]. However, the phylogenetic analysis of
Theileria using 28S rRNA has not been reported previously.
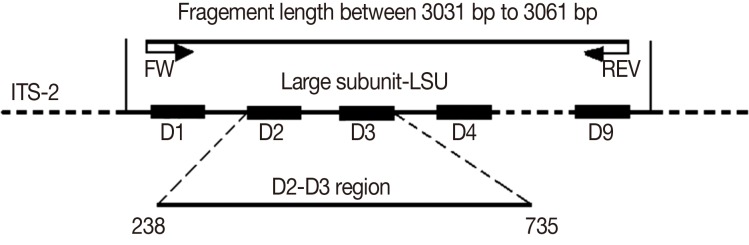
The 28S rRNA gene comprises a mixture of conserved and divergent regions. The divergent regions of the 28S rRNA gene are known as "D"-regions and are numbered from the 5' to 3' direction in the rRNA [
15,
19] (
Fig. 1). The D2 and D3 segments of the 28S rRNA are usually selected in phylogenetic studies because it is easy to design primers for them and there are many informative sites [
20]. Therefore, an increasing number of studies have moved from 18S rRNA to the D2 and D3 segments of 28S rRNA to analyze phylogenetic relationships [
21-
24].
In this study, we obtained 28S rRNA sequences and D2-D3 fragments from 13 Theileria spp. isolates collected in China. The phylogenetic analysis was performed using the maximum parsimony (MP) and Bayesian inference (BI) methods to determine the relationships among Theileria spp.
MATERIALS AND METHODS
Parasites and animals
Thirteen Chinese isolates of
Theileria were used in this study, which comprised 9 isolates infective to cattle, i.e.,
T. orientalis Ningxian,
T. orientalis Wenchuan,
T. orientalis Lintan,
T. sinensis Lintan,
T. sinensis Weiyuan,
T. annulata Ningxia,
T. annulata Sanmenxia,
T. annulata Inner Mongolia, and
T. annulata Xinjiang, and 4 isolates infective to sheep, i.e.,
T. ovis Xinjiang,
T. uilenbergi Longde,
T. luwenshuni Weiyuan, and
T. luwenshuni Ninxian. Detailed information on the 13 isolates is shown in
Table 1.
Experimental animals (cattle and sheep) aged 6-12 months were purchased from an area where theileriosis had not been reported. One month before the experiments, all animals were splenectomized. Ten days before the experiment, blood films were taken from the ears of the animals, which were stained with Giemsa and examined to confirm the absence of hemoparasites [
25]. The animal experiments were approved and performed according to the guidelines of Institutional Animal Care and Use Committee, and the number of IACUC is SYXK2010-0003.
Nine cattle and 4 sheep were inoculated with 10 ml of cryopreserved infectious blood containing different
Theileria isolates. When the parasitemia level reached >5%, blood was drawn from the jugular vein and collected in tubes using heparin as an anticoagulant. Erythrocytes were isolated and the parasite DNA was obtained using a DNA MiniKit (QIAGEN GmbH, Hilden, Germany), according to the manufacturer's instructions. The amount of DNA isolated was assessed photometrically. Negative control DNA was obtained from the experimental animals prior to inoculation [
26].
A pair of 28S rRNA sequence piroplasma universal primers were used based on the sequences of
T. parva (GenBank no. AF218825 and no. AF013419) and a previous study [
25]. The primers were as follows: forward, 5'(1011)-CTAGTAACG-GCGAGCGAAGA-3'(1030); reverse, 5'(4056)-AGGCGTTCAGTCATTATCCAA-3'(4036). The numbers in parentheses indicate the nucleotide positions of the consensus sequence. The PCR amplification conditions and the generation of sequences used the same method as a previous study [
25]. The 18S rRNA sequence of each isolate was downloaded from the GenBank (
Table 1).
Contigs were assembled using the Lasergene SeqMan, and the sequences produced were checked against GenBank to verify that the sequences were from the appropriate
Theileria species. The sequences were initially compared with public sequences using the BLAST program at NCBI, USA (
http://www.ncbi.nlm.nih.gov/). The distance matrices for the aligned sequences, where all gaps were ignored, were calculated using the Kimura's 2-parameter method in MEGA4. The D2-D3 segments with junction sequences from base pair positions 238-735 were selected based on the alignment results of 15 isolates.
Phylogenetic trees of the complete 28S rRNA, D2-D3 region, and 18S rRNA were constructed using the maximum parsimony (MP) and Bayesian inference (BI). The MP trees were constructed using the PAUP4.0 [
27]. Gaps were treated as missing data, with equal weightings for transitions and transversions, and a heuristic search was made using the TBR branch-swapping algorithm [
28]. BI analyses of each dataset were conducted separately using the MrBayes 3.1.2 [
29]. The Bayesian analyses were initiated using a GTR+I+Γ model with no initial values assigned to these parameters and were performed with 4 chains of 1.0×10
6 generations for the 28S rRNA dataset, 2.0×10
5 generations for the D2-D3 dataset, and 1.0×10
6 generations for the 18S rRNA dataset. The Markov chains were sampled at intervals of 100 generations. After discarding the burn-in samples and evaluating convergence, the remaining samples were retained for further analysis. The posterior probabilities (PP) were obtained for appropriate clades. Clades with PP ≥ 95% were considered to be highly supported. One
Babesia bovis isolate was used as the outgroup. Finally, the phylogenetic trees constructed using the 2 methods were viewed using the TreeView program (version 32).
RESULTS
rRNA gene sequences
The lengths of the PCR products of the 28S rRNA gene ranged from 3,037 bp (T. annulata Inner Mongolia) to 3,061 bp (T. orientalis Wenchuan and T. orientalis Lushi). In the aligned sequences of the 28S rRNA gene, 526 sites were variable and 399 sites were parsimony-informative according to the criterion of the maximum parsimony. The nucleotide frequencies were 0.253 (A), 0.262 (T), 0.207 (C), and 0.278 (G). The transition/transversion rate ratios were 2.609 (purines) and 1.417 (pyrimidines). The percentage sequence divergence ranged from 0.1% to 5.8% at the intraspecies level, and from 1.2% to 11% at the interspecies level. The lengths of the selected D2-D3 segments varied from 467 bp (T. annulata Inner Mongolia) to 485 bp (T. orientalis Wenchuan). The percentage of sequence divergence ranged from 0.2% to 4.7% at the intraspecies level, and from 4.4% to 28% at the interspecies level. During this region, 192 sites were variable, and 141 sites were parsimony-informative. The nucleotide frequencies were 0.263 (A), 0.286 (T), 0.194 (C), and 0.256 (G). The transition/transversion rate ratios were 1.526 (purines) and 3.438 (pyrimidines). For 18S rRNA, the variation of sequences ranged from 0.1% to 1.2% at the intraspecies level, and from 1.6% to 4.0% at the interspecies level.
Phylogenetic relationships of Theileria spp.
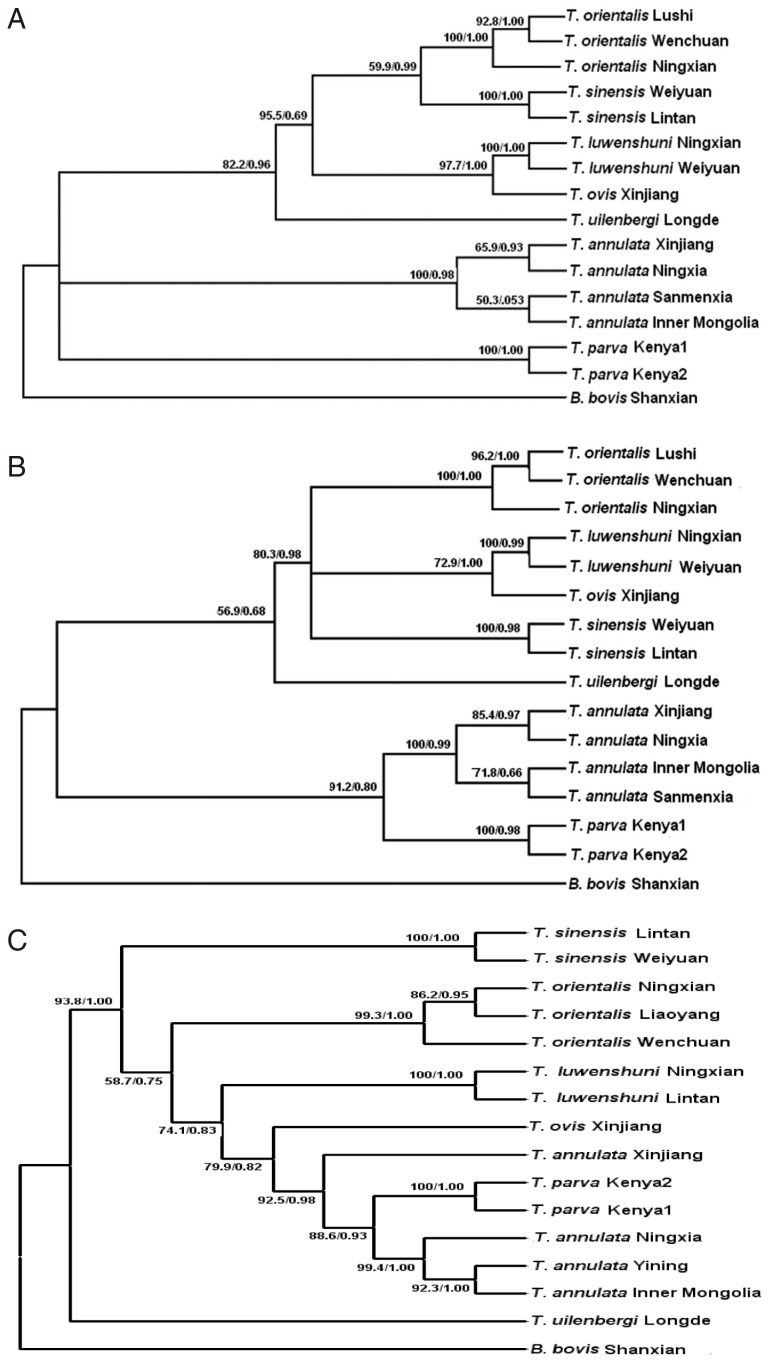
To compare the differentiation of different species of
Theileria, phylogenetic trees were constructed for the 28S rRNA gene, D2-D3 region, and 18S rRNA gene (
Fig. 2A-C). The Bayesian analysis produced essentially the same topology as the maximum parsimony analyses, so the 3 trees generated by PAUP are shown here with the bootstrap proportions (BP) and the posterior probabilities (PP) produced by the Bayesian analyses.
Fig. 2A is based on the 28S rRNA gene and it shows that the 15
Theileria isolates were divided into 3 clades; 1 clade for
T. annulata, another clade for
T. parva, while the remaining
Theileria spp. constituted the remaining clade. The phylogenetic tree generated for the D2-D3 region (
Fig. 2B) showed that
T. annulata and
T. parva constituted 1 clade. However, the biggest difference in the phylogenetic tree based on the 18S rRNA gene was that
T. uilenbergi represented 1 clade and the remaining
Theileria spp. constituted another clade (
Fig. 2C).
DISCUSSION
In this study, we used 1 pair of piroplasma universal primers for 28S rRNA, which has been shown to amplify the DNA of
Babesia spp. and
Theileria spp. successfully [
25]. The amplification efficiency was the same for the 2 piroplasma genera but the lengths of the amplified fragment were different for
Babesia (2,878-3,017 bp) and
Theileria (3,037-3,061 bp) [
25]. This shows that different indel events have occurred in each species of piroplasma. However, its high amplification efficiency means that this pair of primers could potentially be used to develop a method for diagnosing piroplasmosis and for use in epidemiological investigations.
The 18S rRNA gene is used most commonly in studies of
Theileria but it has a high degree of conservation and does not always discriminate among closely related species [
30,
31]. The 28S rRNA gene comprises tandem conserved and diverse regions. Given these structural characteristics, several recent molecular phylogeny studies have focused on 28S rRNA [
32,
33]. Some studies have proved the "D"-regions (especially the D2-D3 segment) are suitable markers for applications in DNA-based species identification and phylogenetic analysis in nematodes and fish [
21,
24,
34]. This is the first study to use the complete sequence of 28S rRNA and the D2-D3 segment as phylogenetic markers to analyze the relationships among
Theileria species. Our results showed that the complete and partial 28S rRNA gene can exhibit better intraspecific diversity and closer interspecific relationships among
Theileria spp. than 18S rRNA. However, from intra-/interspecific distance and phylogenetic relationships, D2-D3 segment has also been proved as a potential marker in
Theileria species.
A comparison of the results of the current study and previous phylogenies obtained using other markers (ITS genetic regions and MPSP) [
11,
14,
35] showed that the biggest difference was the phylogenetic relationships among 3 ovine
Theileria (
T. luwenshuni,
T. uilenbergi, and
T. ovis) isolated from the northwest region of China.
T. luwenshuni and
T. uilenbergi are considered highly pathogenic, whereas
T. ovis is considered to be benign because it causes subclinical infections in small ruminants [
4,
36].
T. luwenshuni and
T. uilenbergi shared the same shape, host, vector tick, pathogenicity, and distribution, but their phylogenetic relationships were distant in the 28S rRNA and D2-D3 segment trees. These results also provided evidence that
T. luwenshuni and
T. uilenbergi are 2 different and valid species [
30,
31].
T. sinensis is a new species that was first isolated from cattle in the Gansu province of China and
Haemaphysalis qinghaiensis was shown to be its vector [
37]. However, it is still uncertain whether this
Theileria isolate should be treated as a new
Theileria species or if it is an existing species. Recent studies compared different genes of
T. sinensis with other
Theileria spp. and found that
T. sinensis was different from known
Theileria species [
14,
38]. In this study, the 3 phylogenetic trees based on 28S rRNA, D2-D3 segment, and 18S rRNA showed that
T. sinensis always formed an obvious clade, which was distant from other
Theileria species. This supports the conclusion that
T. sinensis should be classified as an independent and valid
Theileria species, which agrees with previous phylogenetic studies based on MPSP and ITS [
39,
40].
The 2 most important species in cattle and water buffalo are
T. parva, as the causative agent of the Corridor disease in cattle in South Africa, and East Coast fever (ECF) in eastern and central Africa, and
T. annulata, which causes tropical theileriosis. In the present study, 2 species formed 1 clade in the phylogenetic trees based on 28S rRNA and 18S rRNA. However, this result was also the same as previous studies which used other molecular markers, such as ITS and MPSP [
39,
40].
In conclusion, our study demonstrated that 28S rRNA was useful for determining the relationships among Theileria species. The D2-D3 segment of the 28S rRNA appears to be a potentially useful target for phylogenetic studies of Theileria spp. The regions investigated in this study will enrich our knowledge of the phylogenetic relationships among Theileria species.
9732010CB530206
NSFC309721823107213031001061
9482010-S04
Key Project of Gansu Province1002NKDA0350801NKDA033
NBCITS.MOACARS-38
Sino-Europe Cooperation, MOST, China, State Key Laboratory of Veterinary Etiological Biology ProjectSKLVEB2008ZZKT019
Notes
-
We have no conflict of interest related with this study.
ACKNOWLEDGMENTS
This study was financially supported by the 973 Program (2010CB530206), NSFC (No. 30972182, No. 31072130, No. 31001061), "948" (2010-S04), Key Project of Gansu Province (1002NKDA035 and 0801NKDA033), NBCITS.MOA (CARS-38), Specific Fund for Sino-Europe Cooperation, MOST, China, State Key Laboratory of Veterinary Etiological Biology Project (SKLVEB2008ZZKT019). The research was also facilitated by EPIZONE (FOOD-CT-2006-016236, ASFRISK (No. 211691), ARBOZOONET (No. 211757) and PIROVAC (KBBE-3-245145) of the European Commission, Brussels, Belgium. We are also grateful to International Science Editing for critical correction of this manuscript.
References
- 1. Levine ND. Erhardorina n. g., Ascogregarina polynesiensis n. sp., Eimeria golemanskii n. sp., Isospora tamariscini n. sp., Gregarina kazumii n. nom., new combinations and emendations in the names of apicomplexan protozoa. J Protozool 1985;32:359-363.
- 2. Bai Q, Liu G, Hen G. An unidentified species of Theileria infective for cattle discovered in China. Trop Anim Health Prod 1997;29:43S-47S.
- 3. Luo J, Lu W. Cattle theileriosis in China. Trop Anim Health Prod 1997;29:4S-7S.
- 4. Yin H, Schnittger L, Luo J, Seitzer U, Ahmed JS. Ovine theileriosis in China: a new look at an old story. Parasitol Res 2007;101:S191-S195.
- 5. Liu Z, Hou J, Bakheit MA, Salih DA, Luo J, Yin H, Ahmed JS, Seitzer U. Development of loop-mediated isothermal amplification (LAMP) assay for rapid diagnosis of ovine theileriosis in China. Parasitol Res 2008;103:1407-1412.
- 6. Yin H, Liu Z, Guan G, Liu A, Ma M, Ren Q, Luo J. Detection and differentiation of Theileria luwenshuni and T. uilenbergi infection in small ruminants by PCR. Transbound Emerg Dis 2008;55:233-237.
- 7. Zhang Z. A general review on the prevention and treatment of Theileria annulata in China. Vet Parasitol 1997;70:77-81.
- 8. Stewart N, Uilenberg G, De Vos A. Review of Australian species of Theileria, with special reference to Theileria buffeli of cattle. Trop Anim Health Prod 1996;28:81-90.
- 9. Kakuda T, Shiki M, Kubota S, Sugimoto C, Brown WC, Kosum C, Nopporn S, Onuma M. Phylogeny of benign Theileria species from cattle in Thailand, China and the USA based on the major piroplasm surface protein and small subunit ribosomal RNA genes. Int J Parasitol 1998;28:1261-1267.
- 10. Jeong W, Yoon S, An D, Cho SH, Lee KK, Kim JY. A molecular phylogeny of the benign Theileria parasites based on major piroplasm surface protein (MPSP) gene sequences. Parasitology 2010;137:241-249.
- 11. Liu Q, Zhou YQ, He GS, Oosthuizen MC, Zhou DN, Zhao JL. Molecular phylogenetic studies on Theileria spp. isolates (China) based on small subunit ribosomal RNA gene sequences. Trop Anim Health Prod 2010;42:109-114.
- 12. Uilenberg G. Theileria sergenti. Vet Parasitol 2011;175:386.
- 13. Chae J, Allsopp BA, Waghela SD, Park J, Kakuda T, Sugimoto C, Allsopp MTEP, Gale Wagner G, Holman PJ. A study of the systematics of Theileria spp. based upon small-subunit ribosomal RNA gene sequences. Parasitol Res 1999;85:877-883.
- 14. Gubbels MJ, Hong Y, van der Weide M, Qi B, Nijman IJ, Guangyuan L, Jongejan F. Molecular characterisation of the Theileria buffeli/orientalis group. Int J Parasitol 2000;30:943-952.
- 15. Long EO, Dawid IB. Repeated genes in eukaryotes. Ann Rev Biochem 1980;49:727-764.
- 16. Mugridge NB, Morrison DA, Heckeroth AR, Johnson AM, Tenter AM. Phylogenetic analysis based on full-length large subunit ribosomal RNA gene sequence comparison reveals that Neospora caninum is more closely related to Hammondia heydorni than to Toxoplasma gondii. Int J Parasitol 1999;29:1545-1556.
- 17. Redmond NE, Raleigh J, van Soest RW, Kelly M, Travers SA, Bradshaw B, Vartia S, Stephens KM, McCormack GP. Phylogenetic relationships of the marine Haplosclerida (Phylum Porifera) employing ribosomal (28S rRNA) and mitochondrial (cox1, nad1) gene sequence data. PLoS One 2011;6:e24344.
- 18. Van Spaendonk RML, Ramesar J, van Wigcheren A, Eling W, Beetsma AL, van Gemert GJ, Hooghof J, Janse CJ, Waters AP. Functional equivalence of structurally distinct ribosomes in the malaria parasite, Plasmodium berghei. J Biol Chem 2001;276:22638-22647.
- 19. Hassouna N, Mithot B, Bachellerie JP. The complete nucleotide sequence of mouse 28S rRNA gene. Implications for the process of size increase of the large subunit rRNA in higher eukaryotes. Nucleic Acids Res 1984;12:3563-3583.
- 20. Subbotin SA, Ragsdale EJ, Mullens T, Roberts PA, Mundo-Ocampo M, Baldwin JG. A phylogenetic framework for root lesion nematodes of the genus Pratylenchus (Nematoda): Evidence from 18S and D2-D3 expansion segments of 28S ribosomal RNA genes and morphological characters. Mol Phylogenet Evol 2008;48:491-505.
- 21. Subbotin SA, Sturhan D, Chizhov VN, Vovlas N, Baldwin JG. Phylogenetic analysis of Tylenchida Thorne, 1949 as inferred from D2 and D3 expansion fragments of the 28S rRNA gene sequences. Nematology 2006;8:455-474.
- 22. Nadler SA, De Ley P, Mundo-Ocampo M, Smythe AB, Patricia Stock S, Bumbarger D, Adams BJ, De Ley IT, Holovachov O, Baldwin JG. Phylogeny of Cephalobina (Nematoda): molecular evi dence for recurrent evolution of probolae and incongruence with traditional classifications. Mol Phylogenet Evol 2006;40:696-711.
- 23. He Y, Subbotin SA, Rubtsova TV, Lamberti F, Brown DJF, Moens M. A molecular phylogenetic approach to Longidoridae (Nematoda: Dorylaimida). Nematology 2005;7:111-124.
- 24. Sonnenberg R, Nolte AW, Tautz D. An evaluation of LSU rDNA D1-D2 sequences for their use in species identification. Front Zool 2007;4:6.
- 25. Gou H, Guan G, Ma M, Liu A, Liu Z, Ren Q, Li Y, Yang J, Chen Z, Yin H, Luo J. Phylogenetic analysis based on 28S rRNA of Babesia spp. in ruminants in China. Exp Appl Acarol 2013;59:463-472.
- 26. Luo J, Yin H, Liu Z, Yang D, Guan G, Liu A, Ma M, Dang S, Lu B, Sun C. Molecular phylogenetic studies on an unnamed bovine Babesia sp. based on small subunit ribosomal RNA gene sequences. Vet Parasitol 2005;133:1-6.
- 27. Swofford D. PAUP 4.0 b10: Phylogenetic analysis using parsimony. Sunderland, MA, USA. Sinauer Associates; 2002.
- 28. Posada D. Using Modeltest and PAUP* to select a model of nucleotide substitution. Curr Protoc Bioinformatics 2003;Chapter 6:Unit 6.5.
- 29. Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003;19:1572-1574.
- 30. Niu Q, Luo J, Guan G, Liu Z, Ma M, Liu A, Gao J, Ren Q, Li Y, Qiu J. Differentiation of two ovine Babesia based on the ribosomal DNA internal transcribed spacer (ITS) sequences. Exp Parasitol 2009;121:64-68.
- 31. Li Y, Guan G, Ma M, Liu J. Theileria ovis discovered in China. Exp Parasitol 2011;127:304-307.
- 32. Rosas-Valdez R, Morrone JJ, García-Varela M. Molecular Phylogenetics of Floridosentis Ward, 1953 (Acanthocephala: Neoechinorhynchidae) Parasites of Mullets (Osteichthyes) from Mexico, Using 28S rDNA Sequences. J Parasitol 2012;98:855-862.
- 33. Brant SV, Pomajbíková K, Modry D, Petrželková K, Todd A, Loker E. Molecular phylogenetics of the elephant schistosome Bivitellobilharzia loxodontae (Trematoda: Schistosomatidae) from the Central African Republic. J Helminthol 2013;87:102-107.
- 34. Subbotin SA, Sturhan D, Vovlas N, Castillo P, Tambe JT, Moens M, Baldwin JG. Application of the secondary structure model of rRNA for phylogeny: D2-D3 expansion segments of the LSU gene of plant-parasitic nematodes from the family Hoplolaimidae Filipjev, 1934. Mol Phylogenet Evol 2007;43:881-890.
- 35. Aktas M, Bendele KG, Altay K, Dumanli N, Tsuji M, Holman PJ. Sequence polymorphism in the ribosomal DNA internal transcribed spacers differs among Theileria species. Vet Parasitol 2007;147:221-230.
- 36. Alani A, Herbert I. Pathogenesis of infection with Theileria recondita (Wales) isolated from Haemaphysalis punctata from North Wales. Vet Parasitol 1988;28:293-301.
- 37. Bai Q, Liu G, Yin H, Zhao Q, Liu D, Ren J, Li X. Theileria sinensis sp nov: A new species of Bovine Theileria-Molecular Taxonomic studies. Xu Mu Shou Yi Xue Bao 2002;33:185-190.
- 38. Yin H, Luo J, Schnittger L, Lu B, Beyer D, Ma M, Guan G, Bai Q, Lu C, Ahmed J. Phylogenetic analysis of Theileria species transmitted by Haemaphysalis qinghaiensis. Parasitol Res 2004;92:36-42.
- 39. Liu A, Guan G, Liu Z, Liu J, Leblanc N, Li Y, Gao J, Ma M, Niu Q, Ren Q, Bai Q, Yin H, Luo J. Detecting and differentiating Theileria sergenti and Theileria sinensis in cattle and yaks by PCR based on major piroplasm surface protein (MPSP). Exp Parasitol 2010;126:476-481.
- 40. Liu J, Guan G, Liu Z, Liu A, Ma M, Bai Q, Yin H, Luo J. Additional data for a new Theileria sp. from China based on the sequences of ribosomal RNA internal transcribed spacers. Exp Parasitol 2012;133:217-221.
Fig. 1Primers used for amplification and sequencing of 28S rDNA and approximate lengths of fragments. The sketch below indicates the positions of the D2-D3 region. The numbers indicate base pair positions of the alignment results for 15 isolates of Theileria spp. used in this study.

Fig. 2Phylogenetic trees based on the complete 28S rDNA gene (A), D2-D3 region (B), and 18S rDNA gene (C) from Theileria spp., which were computed using the maximum parsimony (MP) and Bayesian inference (BI) algorithms. Clades with bootstrap (BP) support and posterior probabilities (PP) are marked at the nodes. One isolate of Babesia bovis was used as the outgroup.
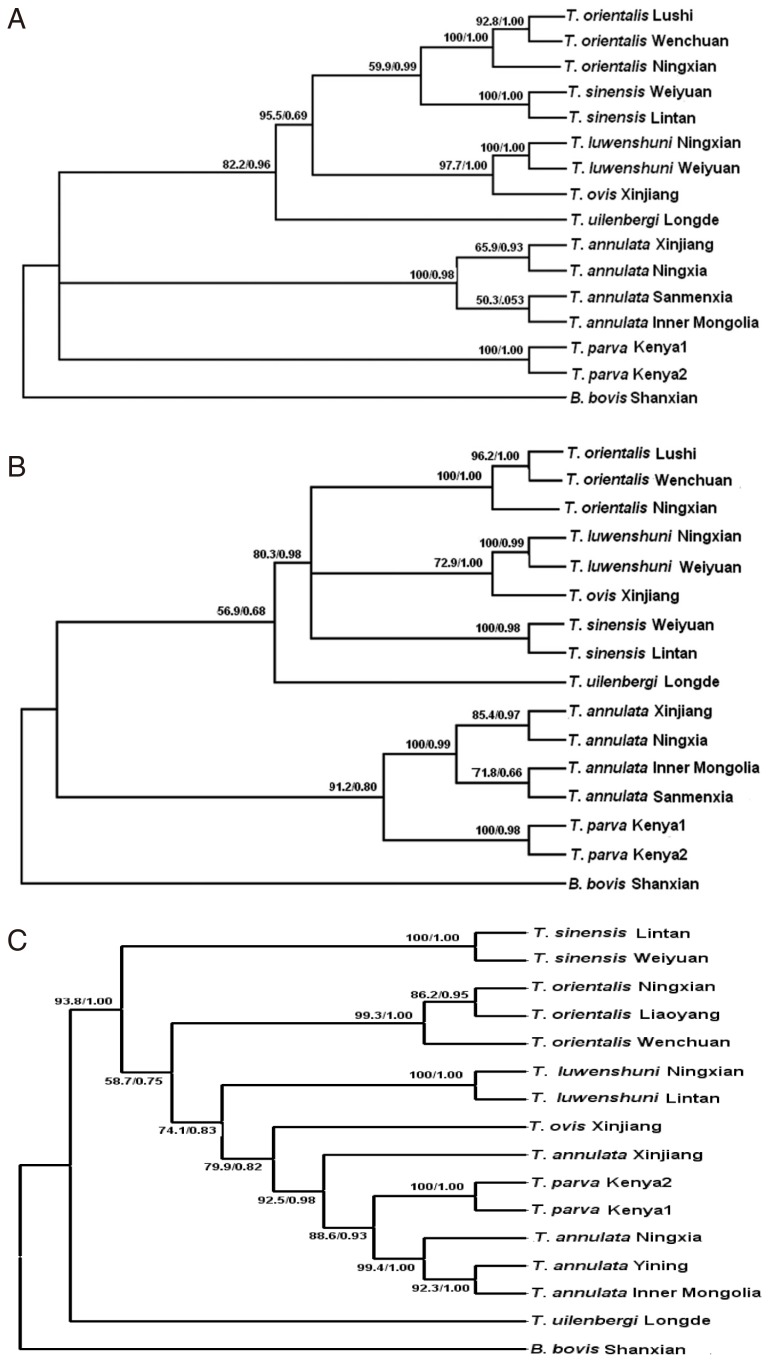
Table 1.Host and origin of Theileria spp., and GenBank accession number of 28S rDNA and 18S rDNA sequences used in phylogenetic analysis
Table 1.
|
Species |
Origin |
Host |
Type |
Length (bp) |
Accession no. |
|
Theileria annulata
|
Sanmenxia (China) |
Cattle |
28S |
3,038 |
JN696675 |
|
Yining (China) |
|
18S |
1,739 |
EU073963 |
|
T heileria annulata
|
Inner Mongolia (China) |
Cattle |
28S |
3,037 |
JN696678 |
|
|
|
18S |
1,740 |
EU083801 |
|
Theileria annulata
|
Xinjiang (China) |
Cattle |
28S |
3,040 |
JN696671 |
|
|
|
18S |
1,742 |
EU083799 |
|
Theileria annulata
|
Ningxia (China) |
Cattle |
28S |
3,050 |
JN696676 |
|
|
|
18S |
1,740 |
EU083800 |
|
Theileria orientalis
|
Ningxian (China) |
Cattle |
28S |
3,060 |
JN696679 |
|
|
|
18S |
1,738 |
EU083803 |
|
Theileria orientalis
|
Wenchuan (China) |
Cattle |
28S |
3,061 |
JN696677 |
|
|
|
18S |
1,876 |
EU083804 |
|
Theileria orientalis
|
Lushi (China) |
Cattle |
28S |
3,061 |
JN696670 |
|
Liaoyang (China) |
|
18S |
1,738 |
EU083802 |
|
Theileria sinensis
|
Lintan (China) |
Cattle |
28S |
3,058 |
JN696681 |
|
|
|
18S |
1,741 |
EU274472 |
|
Theileria sinensis
|
Weiyuan (China) |
Cattle |
28S |
3,057 |
JN696673 |
|
|
|
18S |
1,738 |
EU277003 |
|
Theileria ovis
|
Xinjiang (China) |
Sheep |
28S |
3,056 |
JN696672 |
|
|
|
18S |
1,744 |
FJ603460 |
|
Theileria luwenshuni
|
Weiyuan (China) |
Sheep |
28S |
3,058 |
JN696680 |
|
Lintan (China) |
|
18S |
1,743 |
AY262115 |
|
Theileria luwenshuni
|
Ningxian (China) |
Sheep |
28S |
3,058 |
JN696669 |
|
|
|
18S |
1,744 |
AY262118 |
|
Theileria uilenbergi
|
Longde (China) |
Sheep |
28S |
3,051 |
JN696674 |
|
|
|
18S |
1,742 |
AY262120 |
|
Theileria parva
|
Kenya1 |
Buffalo |
28S |
3,268 |
AF218825 |
|
|
|
18S |
1,740 |
L02366 |
|
Theileria parva
|
Kenya2 |
Buffalo |
28S |
3,294 |
AF013419 |
|
|
|
18S |
1,739 |
AF013418 |
|
Babesia bovis
|
Shanxian (China) |
Buffalo |
28S |
2,878 |
JN391431 |
|
|
|
18S |
1,653 |
AY603398 |