Abstract
The vir genes are antigenic genes and are considered to be possible vaccine targets. Since India is highly endemic to Plasmodium vivax, we sequenced 5 different vir genes and investigated DNA sequence variations in 93 single-clonal P. vivax isolates. High variability was observed in all the 5 vir genes; the vir 1/9 gene was highly diverged across Indian populations. The patterns of genetic diversity do not follow geographical locations, as geographically distant populations were found to be genetically similar. The results in general present complex genetic diversity patterns in India, requiring further in-depth population genetic and functional studies.
-
Key words: Plasmodium vivax, vir gene, DNA sequence polymorphism, India
Plasmodium vivax is the most widely spread human malaria parasite and is a major cause of concern in the Central and South America, Central, South and Southeast Asia, India, Middle East, Oceania, and East Africa. Historically, India is highly endemic to
P. vivax but recent reports suggest that malaria due to
Plasmodium falciparum and
P. vivax are in equal proportions [
1]. The portrayal of
P. vivax as benign is now being challenged, as reports of respiratory distress and coma have emerged along with the increasing resistance of the parasite to chloroquine [
2,
3,
4,
5,
6]. This situation complicates the epidemiology as well as outcome of
P. vivax malaria not only in India but in global context calling for the urgent attention for effective control measures.
Antigenic genes are present in malaria parasites which encode the variant surface antigens (VSAs) providing protective immunity to the parasite against host/vector. Among these genes, the
vir family of
P. vivax is the largest subtelomeric multigene superfamily containing 346 genes divided into 12 subfamilies (A-L) [
7,
8]. The function of the
vir gene superfamily is not yet clearly known but they are likely to be involved in antigenic variation and cytoadherence [
9,
10]. The virulent nature of the VIR proteins leads us to believe that they have a potential role in malaria pathogenesis [
7]. Therefore, it is important to study the population genetic diversity of this gene family to understand the recent changing trends of
P. vivax.
Indian
P. vivax displays complex evolutionary history and holds several traits of being part of the ancestral distribution range [
11]. Not many population genetic studies have been carried out on the
vir genes of
P. vivax in India. However, 4
vir genes were analyzed for the genetic variability existing in different
P. vivax populations in India and were found to be highly divergent [
12]. In this study, these
vir genes were further analyzed by population genetic approaches using different statistical tools to facilitate the understanding of the existing diversity of this superfamily among different Indian populations.
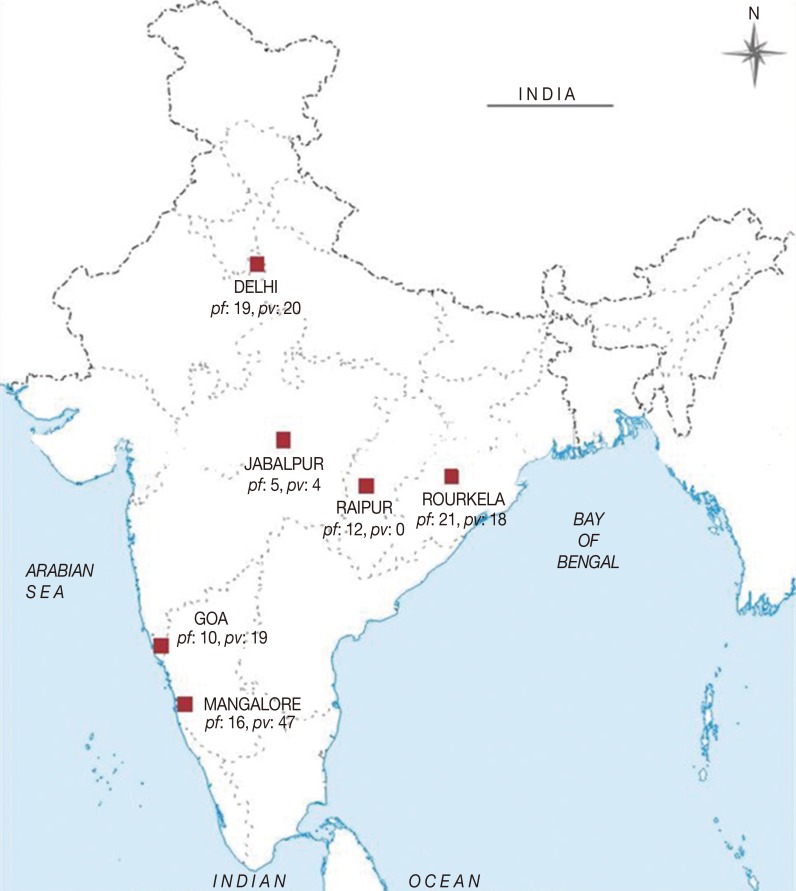
In the present study, a total of 191 malaria symptomatic blood samples were collected from patients by finger-prick method. The samples were collected from 6 different epidemiological settings in India, i.e., 39 from Delhi (DEL) in north India, 63 from Mangalore (MNG) in south India, 29 from Goa (GOA) in west India, 39 from Rourkela (RKL) in east India, 9 from Jabalpur (JBL), and 12 from Raipur (RPR) in central India during the years 2008-2011 (
Fig. 1).
The ethical clearances for the proposed study to collect blood samples were obtained from the Institutional Ethics Committee, National Institute of Malaria Research (NIMR), New Delhi, India. The patients were briefed about the study verbally and provided a written consent before the samples were collected. Preliminary diagnosis of
P. vivax was done by microscopy followed by rapid diagnostic tests (RDT) (Bioline SD Rapid Test, San Diego, California, USA). The bloodspots were made on Whatman (no. 3) filter paper strips for further molecular studies and transported to the lab in air-tight sealed bags. Genomic DNA of
P. vivax was extracted by QIAamp DNA Blood Mini Kit (Qiagen Inc., Valencia, California, USA) according to the manufacturer's instructions. The isolates were analyzed for mixed infections by
P. falciparum and
P. vivax with PCR assays using the published 18s rRNA primers [
13]. Furthermore, the single clones of
P. vivax were determined by genotyping the isolates with merozoite surface protein 3α (
msp3α) gene.
We followed the traditional PCR protocol using the primers as described earlier for
vir 27,
vir 4,
vir 12, and
vir 21 [
12]. The data generated during the study of these 4 genes have already been published [
12]. In this study, we have included 1 more novel
vir gene (
vir 1/9) and have reanalyzed the data of the 5
vir genes (4 previously published and 1 described in the present study) following different statistical tools to infer diversity patterns among these 5
vir genes. The 5
vir genes belonged to different subfamilies, i.e.,
vir 27 in subfamily I,
vir 4 in subfamily C,
vir 12 in subfamily E,
vir 21 in subfamily B, and
vir 1/9 in subfamily J [
2]. The
vir 1/9 gene is 871 bp in length and comprises of 2 exons and 1 intron. Considering the second exon of the
vir genes to be highly variable [
14], we have designed novel primers to amplify the second exon of the
vir 1/9 gene.
The sequences of the primers are; v1/9_1: 5' ATGACA-AATGGGGGACTCAA 3' (forward) and v1/9_3: 5' GAAAATTACTGTTTCCTTAAAATGTGT 3' (reverse) for the primary PCR reaction, and v1/9_2: 5' CGTGAAATGTTATCGGAAAATG 3' (forward) and v1/9_3 for the semi-nested PCR reaction. The annealing temperature was 50℃. The purified PCR products were sequenced as described earlier [
12]. The accession numbers of the 4
vir genes (
vir 27,
vir 4,
vir 12, and
vir 21) are available in GenBank under accession nos. JQ733915-JQ733988 [
12]. The homologous sequences of all the 5
vir genes were compared with the Sal-I reference sequences with GenBank nos. (AAKM-01000041.2 for
vir 27, AAKM01000104.1 for
vir 4, AAKM-01000016.1 for
vir 12, AAKM01000003.1 for
vir 21, and AAKM-01000050.1 for
vir 1/9).
The sequenced DNA fragments for each
vir gene were viewed in Finch TV computer program and, edited DNA sequences were then aligned separately to detect single nucleotide polymorphisms (SNPs) with the help of MEGA v 5.10 computer program following the ClustalW algorithm [
15]. Minimum evolution phylogenetic tree was constructed for
vir 1/9 from the sequences of the isolates and Sal-I reference sequence. The computer program DnaSP v 5.10 was used for the sequence analysis of the
vir genes [
16]. For each gene and population sample, the number of segregating sites, number of haplotypes, haplotype diversity, and 2 different measures of nucleotide diversity, π and θ
w, were calculated [
17]. Both π and θ
w were used to estimate the extent of nucleotide diversity in a population independently for each of the 5 different
vir genes. Whereas π measures the average number of pairwise nucleotide differences in a set of DNA sequences, θ
w measures the total number of segregating sites in a set of DNA sequences [
18,
19]. Tajima's
D test of neutrality [
20] which compares the number of segregating sites per site with the nucleotide diversity was conducted for each gene and each population, and the
D values were calculated. The direction of Tajima's
D test can provide useful information about the evolutionary forces that a population has undergone. For example, a negative value of Tajima's
D highlights an excess of low frequency polymorphisms which shows population size expansion and purifying selection and a positive value signifies low levels of both low and high frequency polymorphisms which shows decreased population size and balancing selection [
21]. All the values were considered significant at
P<0.05. Furthermore, Pairwise Nei's genetic distances (D) were calculated for the 4
vir genes (
vir 27,
vir 12,
vir 21, and
vir 1/9) independently using GenAlEx v 6.5. For each gene, the population pair-wise genetic distance matrix was used to construct Neighbor-Joining (NJ) phylogenetic trees using the MEGA v 5.10 computer program [
22].
Following the preliminary diagnosis by microscopy and RDT of 191 malaria symptomatic patients, only 108 samples were found to be infected with
P. vivax and the remaining 83 with
P. falciparum. Further confirmation of differential
P. falciparum and
P. vivax infections came from the PCR diagnosis by a nested 18s rRNA PCR assay. However, with PCR assays, 15 isolates from Mangalore were detected as mixed malaria infections with both
P. falciparum and
P. vivax. Therefore, we discarded the 15 samples from further analyses. The remaining 93
P. vivax single infections were further found to be single clonal with
msp3α and thereafter analyzed with
vir specific primers. The distribution of isolates in each Indian population is depicted in
Fig. 1.
The amplified sequence lengths for the
vir genes of the present study ranged between 258-1,314 bp. The sequenced DNA fragments of the 5
vir genes were independently aligned (with the respective reference sequences of the SAL-1 strain), manually edited and all the insertions as well as deletions were removed. Multiple sequence alignment of
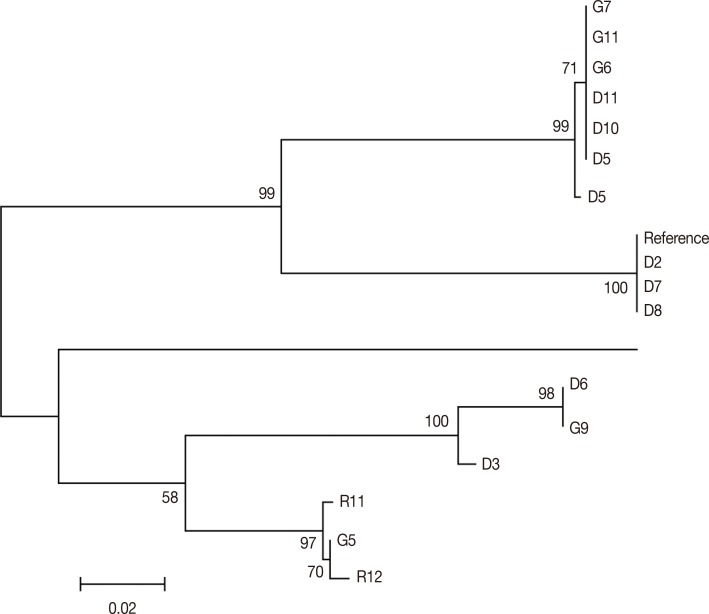
vir 1/9 gene showed 78 SNPs out of which only 3 were synonymous mutations showing high diversity in comparison with the Sal-I reference sequence. The NJ phylogenetic tree for 15 samples showed the presence of 2 principal clades. While the first clade comprises of 9 samples (comprising of
P. vivax isolates from DEL and GOA and the reference isolate), the second clade consists of 7 isolates (with isolates from DEL, GOA, and RKL;
Fig. 2). Each clade was further divided into a number of subclades. There was no geographical clustering among the isolates, and the distribution was observed to be randomly presenting very similar profiles as observed in the previous
vir genes [
12]. Comparison of the number of segregating sites among the 5
vir genes revealed the presence of as low as 2 SNPs (
vir 4) to as high as 179 (
vir 21) among Indian
P. vivax populations. Among the populations, the sample from GOA had less segregating sites for
vir 27,
vir 12, and
vir 1/9 as compared to the other populations and maximum number of segregating sites for
vir 21. Similarly, the number of haplotypes varies among the 5 different
vir genes, with the lowest in the
vir 4 (3) and highest in
vir 12 (17). Furthermore, the number of haplotypes of
vir genes in different populations varies from 1-8 (
Tables 1,
2). Very similarly, the haplotype diversity was the lowest in
vir 4 (0.711) and the highest in
vir 12 (0.962) (
Table 1).
The nucleotide diversity parameters as measured by π and θ
w were calculated separately for each gene and population. The average nucleotide diversity parameter π for all 5 genes was 0.047142, and the average value of θ
w was 0.03686 (
Table 1). The average value of π was found to be higher than θ
w in
vir 12,
vir 21, and
vir 1/9, but lower in
vir 27 and almost equal in
vir 4 gene. The π and θ
w estimates were highest in
vir 1/9 (π=0.11280 and θ
w=0.08805) and lowest in
vir 4 (π=0.00068 and θ
w=0.00054) (
Table 1;
Fig. 2). Among all the 6 populations, GOA had the lowest nucleotide diversity in
vir 27 and
vir 12 genes. In general, the pattern of nucleotide diversities as estimated by π and θ
w were quite variable across the 6 populations of
P. vivax (
Table 1), indicating high diversity among the
vir genes in Indian populations. Very similar to the estimates of nucleotide diversity, the Tajima's
D values were quite variable across populations and among the
vir genes (
Tables 1,
2). Wide variations in the Tajima's
D values therefore indicate ongoing molecular evolution of the
vir genes in the Indian
P. vivax populations.
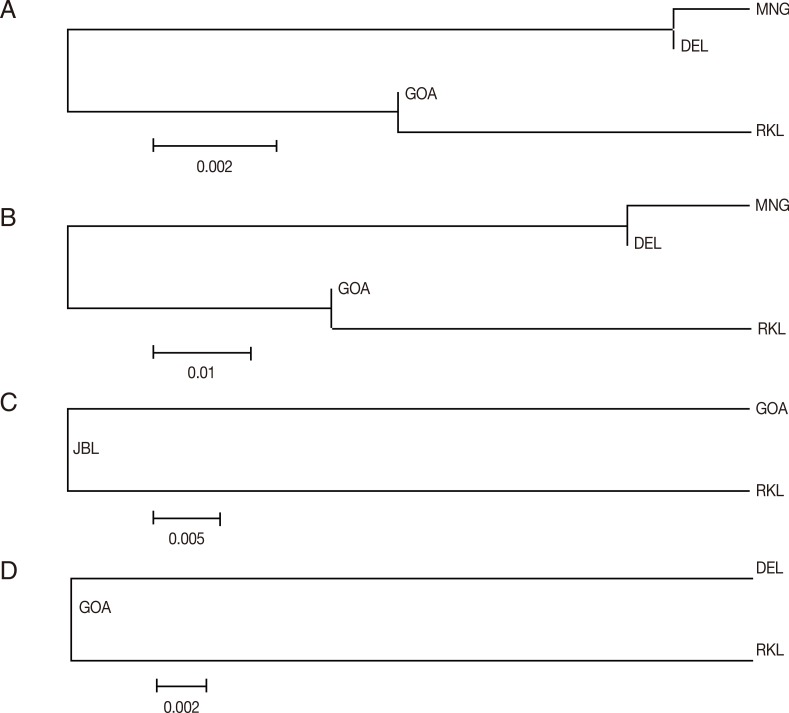
In order to infer genetic interrelationships among Indian population samples of
P. vivax with respect to 4
vir genes sequenced earlier [
12], NJ trees were constructed based on the population pair-wise genetic distance matrix (see above). As shown in
Fig. 3A-D, the placement of Indian population samples in the NJ phylogenetic tree was different for different
vir genes. For example, in the NJ trees constructed with the
vir 27 and
vir 12 genes, DEL and MNG populations come in 1 cluster and GOA and RKL populations in another, as observed with the
vir 1/9 gene (see above). The placement of different Indian populations in the NJ phylogenetic tree majorly points towards no particular patterns of genetic relatedness among the Indian populations of
P. vivax, as for each
vir gene very different patterns were observed. The results therefore in one hand corroborate the earlier finding on no geographic sub-structuring of Indian
P. vivax [
7], on the other hand, high sequence diversity of the
vir genes as previously reported in India [
12] has also been found for the
vir 1/9 gene in India.
It is hypothesized that
vir genes have a role in malaria pathogenesis [
7,
23], and
P. vivax uses the high sequence diversity in these genes to gain high virulence. Therefore, the study on the genetic diversity and evolutionary potentiality of the
vir genes is essential to understand malaria transmission, disease severity, vaccine development, and various evolutionary aspects of malaria. Population genetic studies further allow us to comprehend the evolutionary history of
P. vivax, and whether different genes are influenced by natural selection across different geographical regions [
21]. Considering that India contributes majorly to the global endemicity of
P. vivax malaria [
1], it is therefore essential to study genetic diversity of
vir genes in Indian populations. The calculated average nucleotide diversities for the 5
vir genes studied here were quite high (π=0.047142; θ
w=0.03686), indicating that Indian
P. vivax populations maintain high genetic diversity [
11]. Interestingly, the GOA population sample contains the least diversity in all aspects of data in comparison to other Indian populations (signifying a more conserved population of
vir genes) as compared to the other regions of India. Similarly, among the 5 different
vir genes studied here, the
vir 4 gene was found to contain the least genetic diversity, indicating this gene might be under the influence of some evolutionary constraint. However, such a conclusion should be taken with caution, as we could sequence a very low number of
P. vivax isolates for this gene. In contrast, due to the observation of a large number of haplotypes and a high haplotype diversity in
vir 12, it can be concluded that the
vir 12 gene is the most diverged gene among the 5 genes studied here.
The NJ phylogenetic trees were constructed from pairwise Nei's genetic distance matix for each of the 4
vir genes. Interestingly, the placement of Indian populations was quite different for each of the
vir genes. For example, in the NJ trees constructed with the
vir 27 and
vir 12 genes, DEL and MNG were placed in a single clade and GOA and RKL in another, signifying close genetic affinity between these 2 populations with respect to the 2
vir genes (
vir 27 and
vir 12). For these 2 genes, GOA and RKL also appear to be genetically identical, as these 2 populations are also placed in a single clade, although geographically these 2 locations are quite wide apart. It is known that genetic relatedness between geographically distant populations can arise owing to the common gene pool shared by the isolates in the past [
11]. This phenomenon seems to be common in
P. vivax populations in India, as very similar patterns of clustering was observed for
vir 21 as well as in
vir 1/9 genes. The overall observation in Indian
P. vivax populations thus revealed that the existing diversity in
vir genes was randomly distributed without any definite geographic pattern. No observation on any genetic structure among populations with differential malaria endemicity further corroborates this contention. Such genetic epidemiological differences spread across the country may also be responsible for the increased complexity of the
P. vivax infections which is distinctly exhibited by
vir genes as observed in this study. This population-based study demonstrates differential levels of diversity in the different geographic regions as also reported in other studies [
12]. Observation on the differential calculated values of the Tajima's D also reflect that majorly the Indian
P. vivax populations present complex demographic history, as observed in an earlier study [
11]. Whatever the case may be population genetic studies with more number of
vir genes and functional analyses will reveal more concrete knowledge on the population evolutionary history [
8] of
vir genes in India.
Due to limited information on the genetic diversity studies in
P. vivax populations (in comparison to
P. falciparum), it is difficult to derive concrete conclusion on the observed genetic diversity of the
vir genes found in the present study. Although several studies on genetic polymorphisms in
P. vivax have been conducted in worldwide populations [
24], genetic diversity studies in Indian
P. vivax are limited to some antigenic genes [
25]. The results from all these studies cannot be compared, as molecular markers are not uniform across countries. Therefore, in order to construct map of global genetic diversity patterns of
vir genes, similar
vir genes across
P. vivax malaria endemic countries are essential. Coupled with functional studies, such a diversity map would inform not only on the extent of extant genetic diversity of
vir genes, but also will help in designing new vaccines for
P. vivax malaria management.
Notes
-
We declare that we have no conflict of interest related to this work.
ACKNOWLEDGMENTS
We would like to thank the Department of Biotechnology (DBT) and the Indian Council of Medical Research (ICMR) for providing us the grant required to carry out all the research work for this paper. The paper has clearance from the National Institute of Malaria Research (NIMR) publication committee.
References
- 1. Singh V, Mishra N, Awasthi G, Dash AP, Das A. Why is it important to study malaria epidemiology in India? Trends Parasitol 2009;25:452-457.
- 2. Carlton JM, Adams JH, Silva JC, Bidwell SL, Lorenzi H, Caler E, Crabtree J, Angiuoli SV, Merino EF, Amedeo P, Cheng Q, Coulson RM, Crabb BS, Del Portillo HA, Essien K, Feldblyum TV, Fernandez-Becerra C, Gilson PR, Gueye AH, Guo X, Kang'a S, Kooij TW, Korsinczky M, Meyer EV, Nene V, Paulsen I, White O, Ralph SA, Ren Q, Sargeant TJ, Salzberg SL, Stoeckert CJ, Sullivan SA, Yamamoto MM, Hoffman SL, Wortman JR, Gardner MJ, Galinski MR, Barnwell JW, Fraser-Liggett CM. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 2008;455:757-763.
- 3. Picot S, Bienvenu AL. Plasmodium vivax infection: not so benign. Med Sci (Paris) 2009;25:622-626.
- 4. Kochar DK, Das A, Kochar SK, Saxena V, Sirohi P, Garg S, Kochar A, Khatri MP, Gupta V. Severe Plasmodium vivax malaria: a report on serial cases from Bikaner in northwestern India. Am J Trop Med Hyg 2009;80:194-198.
- 5. Tjitra E, Anstey NM, Sugiarto P, Warikar N, Kenangalem E, Karyana M, Lampah DA, Price RN. Multidrug-resistant Plasmodium vivax associated with severe and fatal malaria: a prospective study in Papua, Indonesia. PLoS Med 2008;5:e128.
- 6. Lacerda MV, Fragoso SC, Alecrim MG, Alexandre MA, Magalhães BM, Siqueira AM, Ferreira LC, Araújo JR, Mourão MP, Ferrer M, Castillo P, Martin-Jaular L, Fernandez-Becerra C, del Portillo H, Ordi J, Alonso PL, Bassat Q. Postmortem characterization of patients with clinical diagnosis of Plasmodium vivax malaria: To what extent does this parasite kill? Clin Infect Dis 2012;55:e67-e74.
- 7. Fernandez-Becerra C, Yamamoto MM, Vêncio RZ, Lacerda M, Rosanas-Urgell A, del Portillo HA. Plasmodium vivax and the importance of the subtelomeric multigene vir superfamily. Trends Parasitol 2009;25:44-51.
- 8. Lopez FJ, Bernabeu M, Fernandez-Becerra C, del Portillo HA. A new computational approach redefines the subtelomeric vir superfamily of Plasmodium vivax. BMC Genomics 2013;14:8.
- 9. Bernabeu M, Lopez FJ, Ferrer M, Martin-Jaular L, Razaname A, Corradin G, Maier AG, del Portillo HA, Fernandez-Becerra C. Functional analysis of Plasmodium vivax VIR proteins reveals different subcellular localizations and cytoadherence to the ICAM-1 endothelial receptor. Cell Microbiol 2012;14:386-400.
- 10. Carvalho BO, Lopes SC, Nogueira PA, Orlandi PP, Bargieri DY, Blanco YC, Mamoni R, Leite JA, Rodrigues MM, Soares IS, Oliveira TR, Wunderlich G, Lacerda MV, del Portillo HA, Araújo MO, Russell B, Suwanarusk R, Snounou G, Rénia L, Costa FT. On the cytoadhesion of Plasmodium vivax-infected erythrocytes. J Infect Dis 2010;202:638-647.
- 11. Gupta B, Srivastava N, Das A. Inferring the evolutionary history of Indian Plasmodium vivax from population genetic analyses of multilocus nuclear DNA fragments. Mol Ecol 2012;21:1597-1616.
- 12. Gupta P, Das A, Singh OP, Ghosh SK, Singh V. Assessing the genetic diversity of the vir genes in Indian Plasmodium vivax population. Acta Trop 2012;124:133-139.
- 13. Gupta B, Gupta P, Sharma A, Singh V, Dash AP, Das A. High proportion of mixed-species Plasmodium infections in India revealed by PCR diagnostic assay. Trop Med Int Health 2010;15:819-824.
- 14. del Portillo HA, Fernandez-Becerra C, Bowman S, Oliver K, Preuss M, Sanchez CP, Schneider NK, Villalobos JM, Rajandream MA, Harris D, Pereira da Silva LH, Barrell B, Lanzer M. A superfamily of variant genes encoded in the subtelomeric region of Plasmodium vivax. Nature 2001;410:839-842.
- 15. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol Biol Evol 2011;28:2731-2739.
- 16. Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009;25:1451-1452.
- 17. Nei M. Molecular evolutionary genetics. USA. Columbia University Press; 1987.
- 18. Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics 1983;105:437-460.
- 19. Watterson GA. On the number of segregating sites in genetical models without recombination. Theor Popul Biol 1975;7:256-276.
- 20. Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989;123:585-595.
- 21. Akey JM, Eberle MA, Rieder MJ, Carlson CS, Shriver MD, Nickerson DA, Kruglyak L. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol 2004;2:e286.
- 22. Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research- an update. Bioinformatics 2012;28:2537-2539.
- 23. Merino EF, Fernandez-Becerra C, Durham AM, Ferreira JE, Tumilasci VF, d'Arc-Neves J, da Silva-Nunes M, Ferreira MU, Wickramarachchi T, Udagama-Randeniya P, Handunnetti SM, del Portillo HA. Multi-character population study of the vir subtelomeric multigene superfamily of Plasmodium vivax, a major human malaria parasite. Mol Biochem Parasitol 2006;149:10-16.
- 24. Chenet SM, Tapia LL, Escalante AA, Durand S, Lucas C, Bacon DJ. Genetic diversity and population structure of genes encoding vaccine candidate antigens of Plasmodium vivax. Malar J 2012;11:68.
- 25. Prajapati SK, Joshi H, Valecha N. Plasmodium vivax merozoite surface protein-3α: a high resolution marker for genetic diversity studies. J Vector Borne Dis 2010;47:85-90.
Fig. 1Map of India showing regions where blood samples of malaria patients were collected. The number of P. falciparum (Pf) and P. vivax (Pv) samples collected from each region are shown in the map.
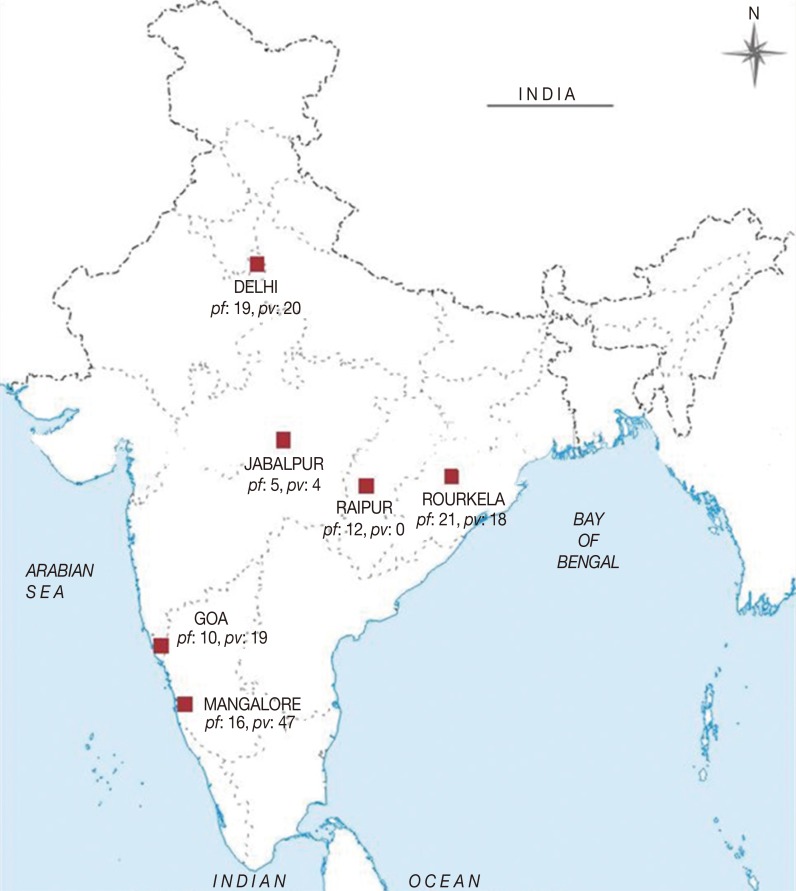
Fig. 2Phylogenetic tree for vir 1/9. The evolutionary history was inferred using the Minimum Evolution method. The optimal tree with the sum of branch length=0.53931989 is shown.

Fig. 3(A) Vir 27 neighbor joining unrooted tree. The phylogenetic tree is based on pairwise Nei genetic distance. The optimal tree with the sum of branch length=0.01525000 is shown. (B) Vir 12 neighbor joining unrooted tree. The phylogenetic tree is based on pairwise Nei genetic distance. The optimal tree with the sum of branch length=0.08350000 is shown. (C) Vir 21 neighbor joining unrooted tree. The phylogenetic tree is based on pairwise Nei genetic distance. The optimal tree with the sum of branch length=0.05150000 is shown. (D) Vir 1/9 neighbor joining unrooted tree. The phylogenetic tree is based on pairwise Nei genetic distance. The optimal tree with the sum of branch length=0.02750000 is shown.
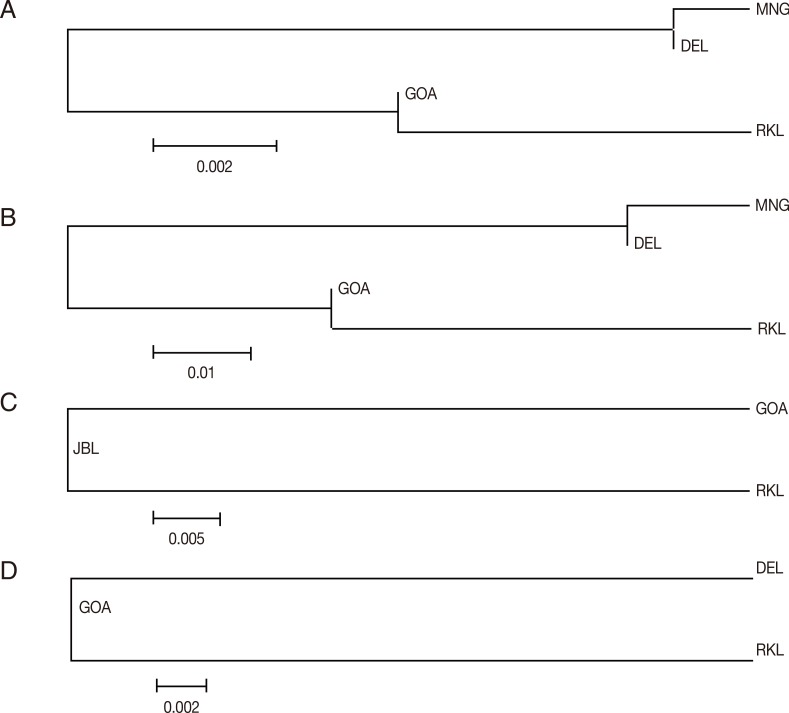
Table 1.Genewise summary statistics for the 5 vir genes
Table 1.
|
Vir genes |
Sequence length |
No. of SNPs (S) |
No. of haplotypes |
Haplotype diversity |
Nucleotide diversity
|
Tests of neutrality
|
|
π |
Θw
|
Tajima’s D |
|
Vir 27 |
1,255 bp |
18 |
15 |
0.845 |
0.00265 |
0.00301 |
-0.36015 |
|
Vir 4 |
1,314 bp |
2 |
3 |
0.711 |
0.00068 |
0.00054 |
0.83017 |
|
Vir 12 |
686 bp |
120 |
17 |
0.962 |
0.06401 |
0.0458 |
1.47617 |
|
Vir 21 |
970 bp |
179 |
13 |
0.925 |
0.05557 |
0.0469 |
0.70136 |
|
Vir 1/9 |
258 bp |
78 |
9 |
0.855 |
0.1128 |
0.08805 |
1.07093 |
|
Average |
|
79.4 |
11.4 |
0.8596 |
0.047142 |
0.03686 |
0.743696 |
Table 2.Population wise summary statistics for the 5 vir genes analyzed
Table 2.
|
Region |
Vir genes |
Sequence length |
No. of segregating sites (S) |
No. of haplotypes |
Haplotype diversity |
Nucleotide diversity
|
Tests of neutrality
|
|
π |
Θw
|
Tajima’s D |
|
Mangalore |
Vir 27 |
1,255 bp |
13 |
6 |
0.833 |
0.00283 |
0.00381 |
-1.22450 |
|
Vir 12 |
686 bp |
101 |
5 |
1.000 |
0.07201 |
0.07697 |
-0.48969 |
|
Delhi |
Vir 27 |
1,255 bp |
5 |
5 |
1.000 |
0.00191 |
0.00191 |
0.00000 |
|
Vir 12 |
686 bp |
91 |
4 |
0.900 |
0.07172 |
0.06647 |
0.59975 |
|
Vir 1/9 |
258 bp |
75 |
6 |
0.929 |
0.12915 |
0.12407 |
0.22226 |
|
Goa |
Vir 27 |
1,255 bp |
3 |
4 |
0.583 |
0.00053 |
0.00088 |
-1.51297 |
|
Vir 12 |
686 bp |
65 |
5 |
1.000 |
0.05015 |
0.04828 |
0.29278 |
|
Vir 21 |
970 bp |
176 |
7 |
0.917 |
0.06446 |
0.06904 |
-0.34406 |
|
Vir 1/9 |
258 bp |
52 |
3 |
0.700 |
0.10349 |
0.09860 |
0.37405 |
|
Rourkela |
Vir 27 |
1,255 bp |
12 |
8 |
0.956 |
0.00397 |
0.00338 |
0.78364 |
|
Vir 12 |
686 bp |
82 |
3 |
0.833 |
0.06487 |
0.06759 |
-0.42015 |
|
Vir 21 |
970 bp |
117 |
4 |
0.900 |
0.05515 |
0.05839 |
-0.42177 |