Abstract
Plasmodium vivax merozoite surface protein-1 (PvMSP1) gene codes for a major malaria vaccine candidate antigen. However, its polymorphic nature represents an obstacle to the design of a protective vaccine. In this study, we analyzed the genetic polymorphism and natural selection of the C-terminal 42 kDa fragment within PvMSP1 gene (Pv MSP142) from 77 P. vivax isolates, collected from imported cases of China-Myanmar border (CMB) areas in Yunnan province and the inland cases from Anhui, Yunnan, and Zhejiang province in China during 2009–2012. Totally, 41 haplotypes were identified and 30 of them were new haplotypes. The differences between the rates of non-synonymous and synonymous mutations suggest that PvMSP142 has evolved under natural selection, and a high selective pressure preferentially acted on regions identified of PvMSP133. Our results also demonstrated that PvMSP142 of P. vivax isolates collected on China-Myanmar border areas display higher genetic polymorphisms than those collected from inland of China. Such results have significant implications for understanding the dynamic of the P. vivax population and may be useful information towards China malaria elimination campaign strategies.
-
Key words: Plasmodium vivax, merozoite surface protein-1, genetic polymorphism, natural selection, Myanmar, China
INTRODUCTION
Malaria is a major infectious disease in the Greater Mekong Subregion (GMS) in Asia. Although there has been a considerable decrease in the incidence of malaria in China [
1], Yunnan Province still has the highest transmission area of vivax malaria in China, particularly in the southern border areas adjacent to Myanmar.
Plasmodium vivax is also the most widely distributed species of all 5 human malaria parasites in Southeast Asia and accounts for 65% of malaria cases in Asia and South America [
2]. More attention is being focused on malaria today than any time since the world’s last efforts to achieve eradication over 40 years ago. The global community is now discussing strategies aimed at dramatically reducing malarial disease burden and the eventual eradication of all types of malaria everywhere. As a consequence,
P. vivax, which has long been neglected and mistakenly considered benign, is now entering into the strategic debates taking place on malaria epidemiology and control, drug resistance, pathogenesis, and vaccines. Thus, contrary to the past, the malaria research community is becoming more aware and concerned about the widespread spectrum of illness and death caused by up to a couple of hundred million cases of vivax malaria each year [
3].
Taking account of the facts above, availability of
P. vivax malaria vaccine is highly desirable. Advanced studies on genetic diversity of the most variable domain of vaccine candidate
P. vivax merozoite surface proteins (PvMSPs) in field isolates of different countries have been carried on and demonstrated that the diversity of MSPs in
P. vivax is presumed be associated to parasite immune evasion and be important for the rationale of malaria vaccine designs [
4,
5]. Since the 42 kDa fragment of
Plasmodium merozoite surface protein-1 (PvMSP1) contains known B- and T-cell cell epitopes, a PvMSP1
42 vaccine antigen may be capable of conferring protection mediated by providing antigen-specific T-cell help for B-cells and antibody production [
6]. Several previous studies have reported the presence of acquired antibodies against the C-terminus part of the protein called PvMSP1
19 or PvMSP1
42 antigens among individuals during natural
P. vivax infections [
7,
8]. Immunological studies performed on animal models have also proved that the PvMSP1
19 or PvMSP1
42 is one of the promising vaccine candidates against asexual stages of the malaria [
9]. Although genetic polymorphisms in the central repeat region of PvMSP1 has been investigated among other countries in Southeast Asia on
P. vivax isolates [
10], the data is not available for the C-terminus region of this antigen from southern border areas adjacent to Myanmar and the inland cases in China.
The present study aimed to identify the genetic diversity and haplotypes of the gene fragment coding PvMSP142 in P. vivax isolates of malaria cases in China-Myanmar border (CMB) areas and Yunnan, Zhejiang, and Anhui province of inland of China. Moreover, the natural selection of the gene fragment coding PvMSP142 was tested in 4 P. vivax populations from CMB areas and inland of China.
MATERIALS AND METHODS
Ethics statement
This study was conducted according to the principles expressed in the Declaration of Helsinki. Blood collections were made with full informed consent of the patients and following institutional ethical guidelines that were reviewed and approved by the ethics committee at National Institute of Parasitic Diseases, Chinese Center for Disease Control and Prevention.
Blood samples, DNA extraction, and purification
Blood samples were obtained from 77 symptomatic and microscopically confirmed
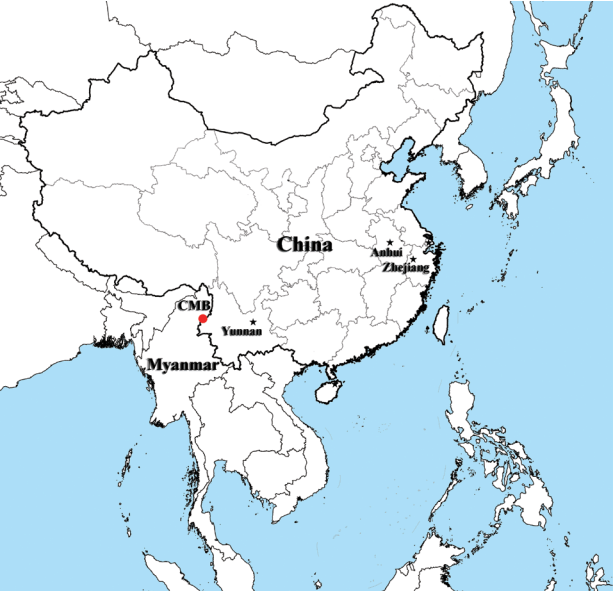
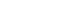
P. vivax malaria patients in China during 2009 to 2012. These examined samples included 59 imported cases of CMB areas based on their traveling history, including 14, 19, 17, and 9 cases each year from 2009 to 2012, respectively, and additional 18 patients from inland China. Among the 18 samples from inland China, 6 of them were from Anhui province collected in 2009, 6 from Zhejiang province collected in 2009 and 6 from Yunnan province collected in 2010 (
Fig. 1). Annual parasite incidence (API) for Anhui, Zhejiang and Yunnan province was 0.0210, 0.0352, and 0.2186 per 10,000 person-years in 2015 [
11]. Because the transmission of malaria had been controlled in a very low level in local China, only limited sporadic cases of malaria inland were collected here. These samples collected from different villages in Yunnan province treated as the inland malaria cases from these febrile patients haven’t been abroad within 1 month. All the patients’
P. vivax infection were diagnosed by microscopic examination of thin and thick blood smears and further confirmed by nested PCR as described previously [
12].
Genomic DNA was isolated from 200 μl of venous blood which collected in a sterile heparinized tube from the patients who were found positive for P. vivax. Approximately 100 μl of blood each patient was used and added 100 μl PBS to get the final volume of 200 μl. Then, DNA was extracted from the whole blood by using the QIAamp DNA mini kit (QIAGEN, Shanghai, China), according to the manufacturer’s instructions. The purified DNA was dissolved in 150 μl TE buffer (10 mM Tris-HCl, 1 mM EDTA; pH 8.0) and stored at −20°C until use.
PCR amplification and analysis of P. vivax field isolates
The
P. vivax fragment (comprising PvMSP1
42 kDa amino acids) of PvMSP1
42 was amplified by polymerase chain reaction (PCR). In this study, the specific primer was designed according to the
P. vivax MSP1 complete gene sequence of PlasmoDB (PVX_099980) [
13]. The primers are: Pv1SF (5′-AGAAG AAAAC GTAGC AGCAA-3′) and Pv1SR (5′-AAGCC CAGTT CAGTT CAGAA CTCA-3′). PCR reaction volumes were 50 μl. The cycling parameters for PCR amplification was performed under the following conditions: initial denaturation 5 min at 94°C, 35 cycles of denaturation at 94°C for 30 sec, annealing at 55°C for 30 sec, and extension at 72°C for 1 min 30 sec, followed by a final extension at 72°C for 5 min. PCR mixture reagents contained 1 μl of DNA, 0.5 units of ExTaq or LATaq DNA polymerase (Takara, Shiga, Japan), 0.2 mM of each primer, 0.1 mM dNTPs in a 25 μl of reaction mix with 1.5 mM MgCl
2. The PCR products were examined by electrophoresis in a 1% agarose gel, visualized with an ultraviolet trans-illuminator and purified with PCR purification kits (Qiagen). Then, the purified PCR products were sequenced using the forward primers on an ABI PRISM 3700 DNA capillary sequencer, by BGI Company (Shenzhen, Guangdong, China). All unique mutations were carefully checked, and ambiguous bases were confirmed by resequencing. We also carried out a BLAST search on PlasmoDB Genebank Database of
P. vivax to compare these successfully sequenced isolates with those previously identified from China and the Asia-pacific subregion. Accurate alignment of the sequences was performed by in ClustalX version 2.0 [
14]. The new sequences were deposited in Gen-Bank with accession nos. JX490129-JX490156, JX993754, and JX993755.
Nucleotide and amino acid sequences were aligned using Clustal W in MEGA 5.0 [
15]. First, nucleotide diversity (π) was computed in 100 bp sliding window and 25 bp step size using DnaSP v.5.0 [
16]. Then, to detect natural selection acting on these coding sequences, the rates of non-synonymous (dN) to synonymous (dS) substitution (dN/dS) was calculated with DnaSP v.5.0. If the amino acid change is deleterious, purifying selection, then dN/dS< 1; only when the amino acid change offers a selective advantage, the dN/dS is> 1. In addition, Tajima’s D was used to test neutrality of this gene fragment in DnaSP v.5.0 [
16,
17]. A remarkable negative value of Tajima’s D reveals an excess of rare variants as expected under positive and negative selection or population size expansion. Whereas, a significant positive value demonstrates an excess of high-frequency variant as expected under balancing selection or under population structure. Finally, to describe the genetic similarities among PvMSP1
42 haplotypes, we constructed networks by the median joining method from 41 unique haplotypes on the basis of PvMSP1
42 sequences in Network 4.5 [
18].
The phylogenetic relationships were derived from the PvM-SP1
42 sequences. In case of individuals that carried an identical sequence (it is possible that individuals from different locations shared the same sequence), only 1 sequence was included for the tree reconstruction. The reference sequences were chosen from GenBank. Then, partitioned Neighbor-Joining method was performed in MEGA 5.0 [
15,
19] to construct the phylogenetic tree (with Kimura-2 parameter distance, branch support with 1000 bootstrap replicates, and complete deletion of gaps). MSP1
42 fragment gene of
P. cynomolgi strain Berok was set as the out group.
RESULTS
Haplotype variations in P. vivax of different field isolates
We successfully amplified and sequenced the gene encoding PvMSP1
42 fragment (1,209 bp, corresponding to amino acid positions 1350–1752 in PvMSP1) shown in
Supplementary Fig. S1. Of 77 isolates from 4 geographic locations, 41 haplotypes were detected on the entire PvMSP1
42 fragment (
Table 1). From these 41 haplotypes, 30 were new haplotypes. Only 1 single haplotype was detected among 6 isolates from Anhui, all these samples were collected from Bengbu city and 2 haplotypes from 6 isolates in Zhejiang province. In contrast, other 6 isolates from Yunnan province distributed in different villages are detected in 6 different haplotypes and 35 haplotypes are detected from 59 imported cases of CMB areas.
Only 3 haplotypes were detected for PvMSP1
19 fragment in comparison with 38 haplotypes for PvMSP1
33 fragment in the all sequenced isolates. The consistent pattern was observed in the
P. vivax populations from CMB areas and inland China (
Table 1).
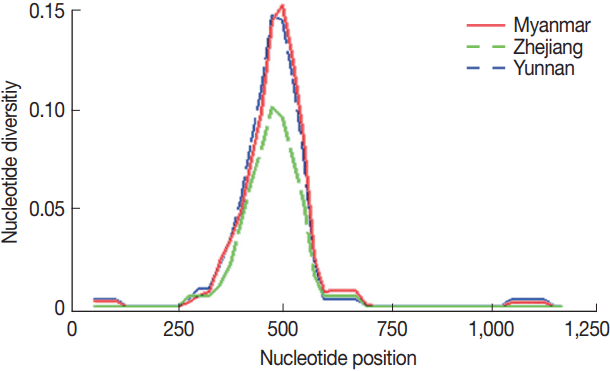
The overall nucleotide diversity (π) of PvMSP1
42 for all of 77 isolates was 0.01901, and π values were 0.01803 and 0.01235 for the isolates from Yunnan and Zhejiang province of inland China, respectively, as well as π value was 0.01836 for the isolates from the CMB areas, with the peak on nucleotide positions from 476 to 525 bp (
Fig. 2), which located at the C-terminal 33 kDa fragment within PvMSP1 gene (PvMSP1
33) (
Supplementary Figs. S2,
S3).
The rates of non-synonymous (dN) to synonymous (dS) substitution (dN/dS) of PvMSP142 for all of 77 isolates was 2.15354, and the rates of dN/dS were 6.10383, 7.39713, and 1.92699 for the isolates from Yunnan, Zhejiang province of inland China and CMB areas, respectively, suggesting a positive selection for PvMSP142 of P. vivax populations from inland China and CMB areas. The overall Tajima’s D value of PvM-SP142 was 2.44824 (P < 0.05) for all of 77 isolates, and the Tajima’s D values were 0.23699, 1.37681, and 2.01030 for the isolates from the Yunnan, Zhejiang province of inland China and CMB areas, respectively, which also indicated balancing selection for PvMSP142 of P. vivax populations from CMB areas.
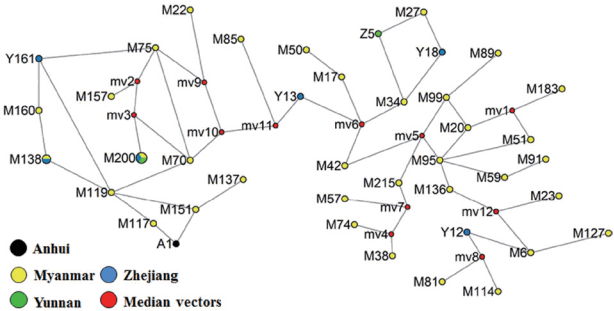
The network of 41 haplotypes from 77 isolates of PvMSP1
42 showed that most prevalent haplotypes originated from Myanmar followed by Yunnan and Zhejiang provinces of China. Moreover, the haplotypes in the studied
P. vivax populations were highly diverse (35/59) in the Myanmar population, and in extremely case, all isolates from Yunnan showed independent haplotypes. In contrast, Zhejiang and Anhui population in inland China showed the low haplotype diversity. The haplotypes were consistent with the distribution of allele frequencies as shown (
Table 1;
Fig. 3).
The overall nucleotide diversity (π) of PvMSP1
33 for all of 77 isolates was 0.02617, and π values were 0.02467 and 0.01716 for the isolates from Yunnan and Zhejiang province of inland China, respectively, as well as π value was 0.02523 for the isolates from the CMB areas (
Table 1;
Fig. 2). The overall nucleotide diversity (π) of PvMSP1
19 for all of 77 isolates was 0.00063, and π values were 0.00098 and 0.00073 for the isolates from Yunnan province of inland China and the CMB areas, respectively (
Table 1;
Fig. 2).
The rates of dN/dS for PvMSP133 were 6.02554, 7.44483, and 1.93351 for the isolates from Yunnan, Zhejiang province of inland China and CMB areas, respectively, suggesting the positive selection located at PvMSP133 fragment of P. vivax populations from inland China and CMB areas. Furthermore, the rate of dN/dS for PvMSP119 was 1.68750 for the isolates from CMB areas.
Moreover, the overall Tajima’s D value of PvMSP1
33 was 2.57163 (
P < 0.05) for all of 77 isolates, and the Tajima’s D values were 0.27447, 1.37681, and 2.11738 for the isolates from Yunnan, Zhejiang province of inland China and CMB areas, respectively, also suggesting a positive balancing selection of PvMSP1
33 in the CMB population (Tajima’s D= 2.11738,
P < 0.05) (
Table 1). However, Tajima’s D values were negative for PvMSP1
19 in the CMB areas (−0.73272) and Yunnan population (−0.93302), indicating the purifying selection (
Table 1).
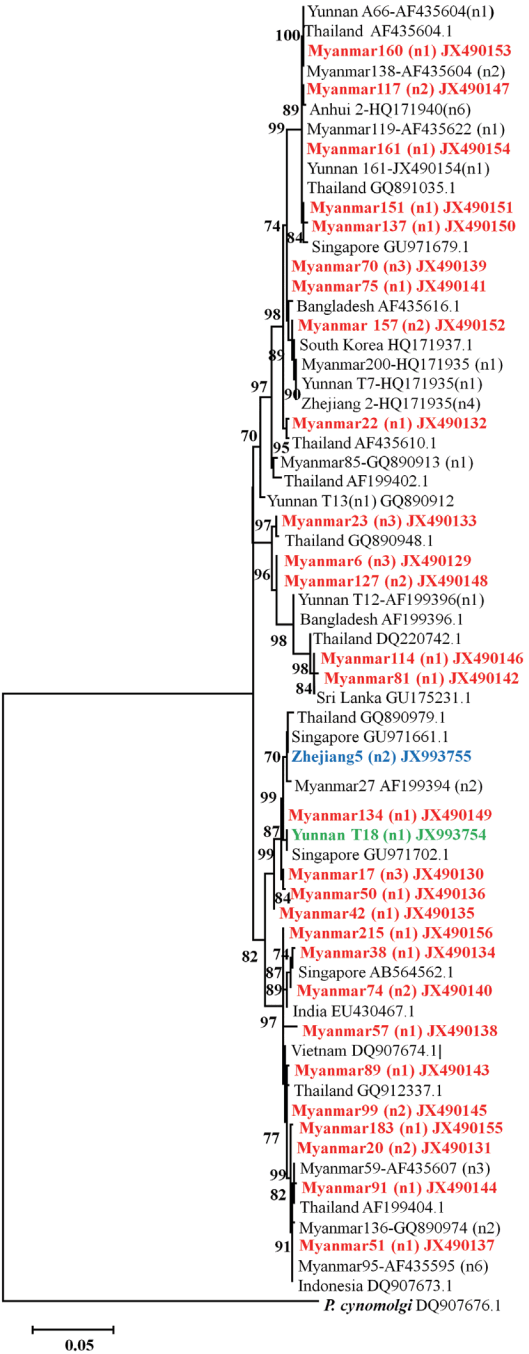
The phylogenic tree showed that all these isolates of
P. vivax clinical patients from different regions such as Myanmar, Thai-land, Singapore, Bangladesh, South Korea, India, Vietnam, and Indonesia having high prevalence (
Fig. 4). What’s more, the
P. vivax clinical samples showed distinct differences haplotypes among the isolates collected from different province in China or different villages in Yunnan province. The phylogenetic analysis revealed that newly identified haplotypes from China were clustered differently. The gene coding PvMSP1
42 from field isolates collected in Anhui province has only 1 haplotype and close to these isolates from Myanmar. Two different haplotypes of PvMSP1
42 were detected from Zhejiang isolates. One sequence coding PvMSP1
42 from Zhejiang province is close to these from Yunnan province and Myanmar; while another one also detected in Zhejiang isolates has been demonstrated to be close to isolates from Singapore and Thailand. All these genes coding PvMSP1
42 from Yunnan are from those patients who haven’t been abroad within 1 month.
DISCUSSION
The
P. vivax parasite exhibits higher genetic diversity than
P. falciparum, especially for the gene families associated with merozoite invasion or immune response modulation (e.g., the
msp3,
vir, and
msp7 gene families) [
20–
22]. The high genetic diversity and natural selection of
P. vivax vaccine targets is common existed in isolates world-wide [
23,
24]. The PvMSP1 locus codes for a major asexual blood-stage antigen currently proposed as a malaria vaccine candidate antigen. Reports of extensive polymorphism of this protein from field isolates and clones from different geographical areas remain a major challenge. Numerous studies on the genetic diversity of PvMSP1 in
P. vivax field isolates have been carried out in many different geographic areas [
25,
26]. However, there is no available data for PvMSP1
42 from southern border areas adjacent to Myanmar and the inland cases in China.
In this study, we present several sets of genetic information for PvMSP1
42 of populations from inland China and CMB areas at first time. We found 35 and 8 haplotypes of PvMSP1
42 for the isolates from Myanmar and China during 2009–2012, respectively. We also documented varied types of haplotypes characteristic of high genetic diversity in the studied region compared to other endemic regions. This high genetic diversity of PvMSP1
42 fragments were consistent with that of
P. vivax field isolates collected in Cambodia and Thailand [
27].
Of the 41 haplotypes, 30 were new haplotypes including 28 of them from Myanmar, characterizing of multiple clonality. The same single haplotype was documented in each of the inland isolates from Anhui compared to those of Myanmar, 2 different haplotypes from isolates from Zhejiang and diverse multiple haplotypes found in Yunnan similar to Myanmar. This finding indicated that geographical proximity between Myanmar and Yunnan China border which showed that vector dynamic and/or human motility might have been important contributing factors in malaria parasite transmission and degree of endemicity [
28]. In recent years, malaria transmission has been controlled in a very low level in Anhui province, China and these cases collected here from Bengbu city were localized sporadic malaria cases [
29]. These results are consistent with previous studies that genetic diversity of the malaria parasites has been shown to be associated with the levels of endemicity and transmission intensity.
Genetic diversity analysis revealed that the majority of polymorphic sites were in the 33 kDa portion and significant proportion of the identified polymorphisms occurred probably as result of reported positive selection pressure on this region while 19 kDa regions remained highly conserved. The similar results for the positive selection of PvMSP1
33 were reported in the
P. vivax isolates from India and Sri Lanka several years ago [
30,
31]. The frequent occurrences of non-synonymous substitutions relative to synonymous ones and high value of Tajima’s D indicate the polymorphism of antigen enable parasites to avoid host immune pressure and host immune responses likely play a role in maintaining the polymorphism of
P. vivax MSP1 alleles.
The halpotype network demonstrated that parasite populations are highly heterogenetic and dynamics of the disease transmission in these endemic areas [
32]. PvMSP1 gene codes for a major malaria vaccine candidate antigen. But its polymorphic nature represents an obstacle to the design of a protective vaccine. Present study will be helpful for the development of PvMSP1 based vaccine against
P. vivax malaria and provide evidence driven knowledge towards development of effective control interventions in Myanmar and appropriate measures in achieving China malaria elimination goals. Noteworthy, Myanmar is one of the major malaria endemic countries in the South-East Asia region, the genetic diversity of the malaria parasite circulating in CMB areas provides additional supportive information. In total, we documented 11 synonymous and 112 non-synonymous haplotypes of which 71.11% and 36.66% previously reported. Of the 11 synonymous polymorphisms, 7 were previously identified. The change might be contributed to evolutionary and/or environmental changes characterized by different patterns compared to natural and geographical studies in the Great Mekong region.
Interestingly, the network analysis of identified haplotypes of PvMSP142 showed that most prevalent haplotypes originated from Myanmar followed by Yunnan and Zhejiang provinces of China. This information is vital and indicates that understanding the genetic diversity and network provides insights into parasite strains dynamics in the region, and design of most appropriate programmes and interventions in reducing or blocking the transmission, curving the spread of parasite as well as containment of increasing resistant strain in the Great Mekong Region.
Phylogenic tree also showed a substantial degree of variability of the origin of the parasites. Although, all
P. vivax clinical isolates, originated from the same species but analysis of these isolates showed distinct differences with the high prevalence of isolates from different countries of Myanmar, Thailand, Singapore, Bangladesh, South Korea, India, Vietnam, and Indonesia and different regions in China. Our findings are consistent with high malaria endemicity in Myanmar, where with the long borders proximity, haplotype diversity has been high comparable to the endemicity in vivax population from inland areas of China such as Anhui and Zhejiang province were lower. However, further studies on a larger population from these endemic geographic areas are required not only to determine the nationwide parasite genetic mapping and detailed malaria molecular epidemiology in CMB areas to provide evidence based decision and effective interventions [
33,
34].
Supplementary materials
Fig. S1
Schematic diagram of Plasmodium vivax merozoite surface protein 1 (PvMSP1). The gene fragment encoding PvMSP142 (including 2 EGF domains and a glycosylphosphatidylinositol anchor) was amplified and sequenced.
Fig. S2
The nucleotide sequence alignment of PvMSP142 from P. vivax isolates collected along the China-Myanmar border areas, local regions of Anhui, Yunnan and Zhejiang provinces in inland China.
Fig. S3
The amino acid sequence alignment of PvMSP142 from P. vivax isolates collected along the China-Myanmar border areas, local regions of Anhui, Yunnan, and Zhejiang provinces in inland China.
Notes
-
CONFLICT OF INTEREST
We have no conflict of interest related to this work.
ACKNOWLEDGMENTS
This work was supported by the National Key Research and Development Program of China (nos. 2016YFC1202000, 2016YFC1202001, 2016YFC1202003 and 2016YFC1200500), the National Natural Science Foundation of China (no. 81101266), the WHA-WHO Demonstration Project (no. UN-OPS/ANDI/G/2016/01), the Foundation of National Science and Technology Major Program (no. 2012ZX10004-220), and the Fourth Round of Three-Year Public Health Action Plan (2015–2017) in Shanghai, P. R. China (no. GWIV-29).
Fig. 1The map of P. vivax samples collection. Sample collection areas in this study are indicated in black pentagrams (Anhui, Yunnan, and Zhejiang provinces from inland China) and red dot (China-Myanmar border area, CMB).
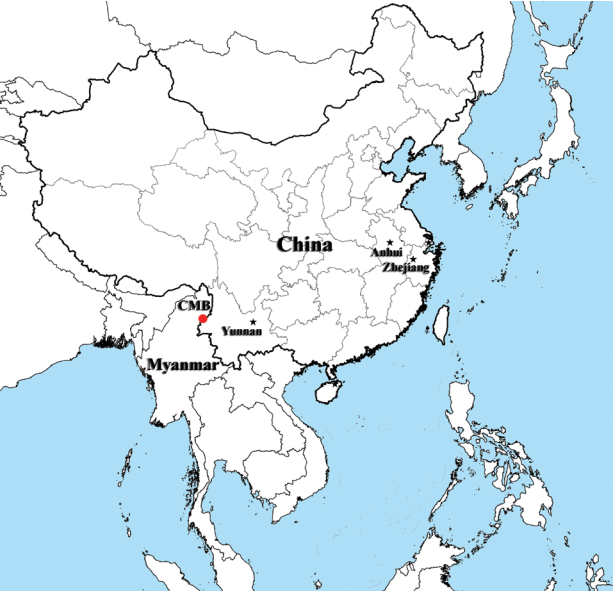
Fig. 2Nucleotide diversity per site (π) at PvMSP142 from P. vivax isolates collected along the China-Myanmar border areas, local regions of Yunnan and Zhejiang provinces in inland China.
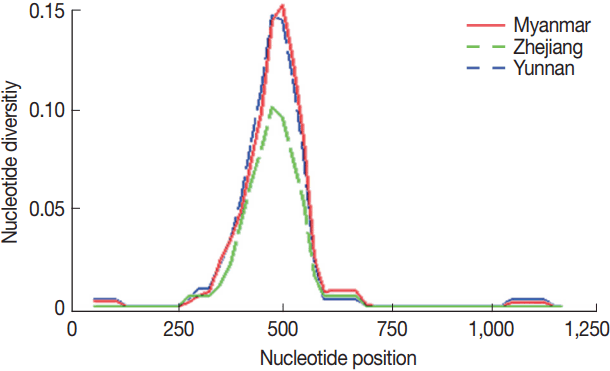
Fig. 3The network of PvMSP142 from P. vivax isolates collected along the China-Myanmar border areas, local regions of Anhui, Yunnan, and Zhejiang provinces in China.
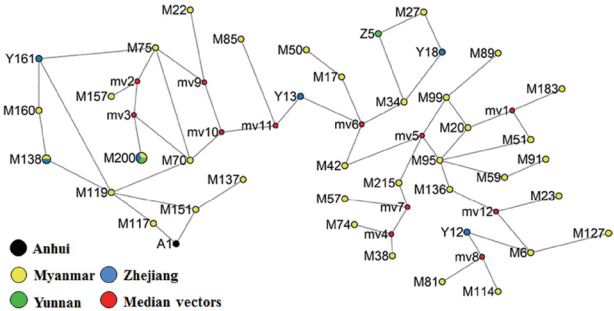
Fig. 4Phylogenetic tree of PvMSP142 from P. vivax isolates collected along the China-Myanmar border areas, local regions of Anhui, Yunnan, and Zhejiang provinces in inland China. Novel sequences identified in this study are indicated in red, blue, and green. Scale bar indicates nucleotide substitutions per site.
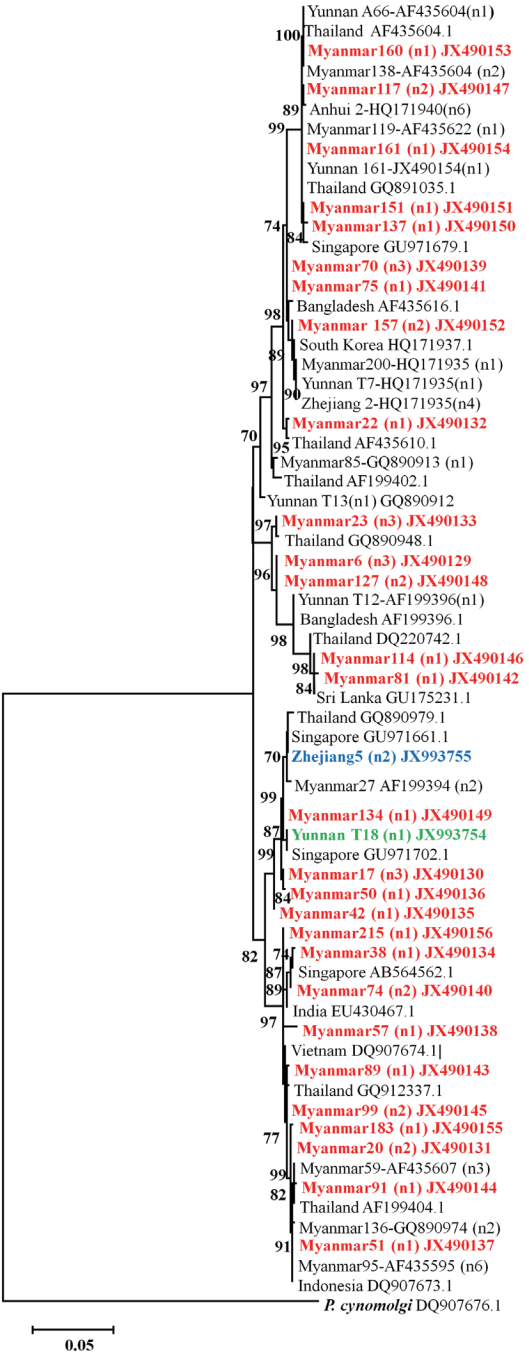
Table 1Halpotype diversity, nucleotide diversity, and natural selection of Plasmodium vivax MSP142
Table 1
|
Fragment |
Ha
|
πb
|
dNc
|
dSd
|
dN/dS |
Tajima’s D |
P-value |
|
42 kDa |
|
All samples (n=77) |
41 |
0.01901 |
0.02188 |
0.01016 |
2.15354 |
2.44824 |
<0.05 |
|
Border areasf (n=59) |
35 |
0.01836 |
0.02085 |
0.01082 |
1.92699 |
2.01030 |
>0.05 |
|
Inland China (n=18) |
8 |
0.01317 |
0.01615 |
−0.00328 |
4.92378 |
0.48590 |
>0.05 |
|
Anhui (n=6) |
1 |
n.ae
|
n.a |
n.a |
n.a |
n.a |
n.a |
|
Yunnan (n=6) |
6 |
0.01803 |
0.02234 |
0.00366 |
6.10383 |
0.23699 |
>0.05 |
|
Zhejiang (n=6) |
2 |
0.01235 |
0.01546 |
0.00209 |
7.39713 |
1.37681 |
>0.05 |
|
|
33 kDa |
|
All samples (n=77) |
38 |
0.02617 |
0.03026 |
0.01401 |
2.15989 |
2.57163 |
<0.05 |
|
Border areas (n=59) |
33 |
0.02523 |
0.02879 |
0.01489 |
1.93351 |
2.11738 |
<0.05 |
|
Inland China (n=18) |
8 |
0.01818 |
0.02237 |
0.00456 |
4.90570 |
0.54855 |
>0.05 |
|
Anhui (n=6) |
1 |
n.a |
n.a |
n.a |
n.a |
n.a |
n.a |
|
Yunnan (n=6) |
6 |
0.02467 |
0.03067 |
0.00509 |
6.02554 |
0.27447 |
>0.05 |
|
Zhejiang (n=6) |
2 |
0.01716 |
0.02159 |
0.00290 |
7.44483 |
1.37681 |
>0.05 |
|
|
19 kDa |
|
All samples (n=77) |
3 |
0.00063 |
0.00071 |
0.00037 |
1.91892 |
−0.76528 |
>0.05 |
|
Border areas (n=59) |
3 |
0.00073 |
0.00081 |
0.00048 |
1.68750 |
−0.73272 |
>0.05 |
|
Inland China (n=18) |
2 |
0.00033 |
0.00042 |
n.a |
n.a |
−1.16467 |
>0.05 |
|
Anhui (n=6) |
1 |
n.a |
n.a |
n.a |
n.a |
n.a |
n.a |
|
Yunnan (n=6) |
2 |
0.00098 |
0.00126 |
n.a |
n.a |
−0.93302 |
>0.05 |
|
Zhejiang (n=6) |
1 |
n.a |
n.a |
n.a |
n.a |
n.a |
n.a |
References
- 1. Yin JH, Zhou SS, Xia ZG, Wang RB, Qian YJ, Yang WZ, Zhou XN. Historical patterns of malaria transmission in China. Adv Parasitol 2014;86:1-19.
- 2. Vogel G. The forgotten malaria. Science 2013;342:684-687.
- 3. Mueller I, Galinski MR, Baird JK, Carlton JM, Kochar DK, Alonso PL, del Portillo HA. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect Dis 2009;9:555-566.
- 4. Wang Y, Ma A, Chen SB, Yang YC, Chen JH, Yin MB. Genetic diversity and natural selection of three blood-stage 6-Cys proteins in Plasmodium vivax populations from the China-Myanmar endemic border. Infect Genet Evol 2014;28:167-174.
- 5. Kassegne K, Abe EM, Chen JH, Zhou XN. Immunomic approaches for antigen discovery of human parasites. Expert Rev Proteomics 2016;13:1091-1101.
- 6. Longley RJ, Sattabongkot J, Mueller I. Insights into the naturally acquired immune response to Plasmodium vivax malaria. Parasitology 2016;143:154-170.
- 7. Chen JH, Chen SB, Wang Y, Ju C, Zhang T, Xu B, Shen HM, Mo XJ, Molina DM, Eng M, Liang X, Gardner MJ, Wang R, Hu W. An immunomics approach for the analysis of natural antibody responses to Plasmodium vivax infection. Mol Biosyst 2015;11:2354-2363.
- 8. Chen JH, Jung JW, Wang Y, Ha KS, Lu F, Lim CS, Takeo S, Tsuboi T, Han ET. Immunoproteomics profiling of blood stage Plasmodium vivax infection by high-throughput screening assays. J Proteome Res 2010;9:6479-6489.
- 9. Dutta S, Kaushal DC, Ware LA, Puri SK, Kaushal NA, Narula A, Upadhyaya DS, Lanar DE. Merozoite surface protein 1 of Plasmodium vivax induces a protective response against Plasmodium cynomolgi challenge in rhesus monkeys. Infect Immun 2005;73:5936-5944.
- 10. Putaporntip C, Jongwutiwes S, Sakihama N, Ferreira MU, Kho WG, Kaneko A, Kanbara H, Hattori T, Tanabe K. Mosaic organization and heterogeneity in frequency of allelic recombination of the Plasmodium vivax merozoite surface protein-1 locus. Proc Natl Acad Sci U S A 2002;99:16348-16353.
- 11. Zhang L, Zhou SS, Feng J, Fang W, Xia ZG. Malaria situation in the People’ s Republic of China in 2015. Chin J Parasitol Parasit Dis 2015;34:477-481. (in Chinese).
- 12. Zhou X, Huang JL, Njuabe MT, Li SG, Chen JH, Zhou XN. A molecular survey of febrile cases in malaria-endemic areas along China-Myanmar border in Yunnan province, People’s Republic of China. Parasite 2014;21:27.
- 13. Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Miller JA, Nayak V, Pennington C, Pinney DF, Roos DS, Ross C, Stoeckert CJ Jr, Treatman C, Wang H. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res 2009;37:539-543.
- 14. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics 2007;23:2947-2948.
- 15. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011;28:2731-2739.
- 16. Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003;19:2496-2497.
- 17. Thornton K. Recombination and the properties of Tajima’s D in the context of approximate-likelihood calculation. Genetics 2005;171:2143-2148.
- 18. Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 1999;16:37-48.
- 19. Zhang W, Sun Z. Random local neighbor joining: a new method for reconstructing phylogenetic trees. Mol Phylogenet Evol 2008;47:117-128.
- 20. Chen SB, Wang Y, Kassegne K, Xu B, Shen HM, Chen JH. Whole-genome sequencing of a Plasmodium vivax clinical isolate exhibits geographical characteristics and high genetic variation in China-Myanmar border area. BMC Genomics 2017;18:131.
- 21. Neafsey DE, Galinsky K, Jiang RH, Young L, Sykes SM, Saif S, Gujja S, Goldberg JM, Young S, Zeng Q, Chapman SB, Dash AP, Anvikar AR, Sutton PL, Birren BW, Escalante AA, Barnwell JW, Carlton JM. The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat Genet 2012;44:1046-1050.
- 22. Shen HM, Chen SB, Wang Y, Xu B, Abe EM, Chen JH. Genome-wide scans for the identification of Plasmodium vivax genes under positive selection. Malar J 2017;16:238.
- 23. Pearson RD, Amato R, Auburn S, Miotto O, Almagro-Garcia J, Amaratunga C, Suon S, Mao S, Noviyanti R, Trimarsanto H, Marfurt J, Anstey NM, William T, Boni MF, Dolecek C, Hien TT, White NJ, Michon P, Siba P, Tavul L, Harrison G, Barry A, Mueller I, Ferreira MU, Karunaweera N, Randrianarivelojosia M, Gao Q, Hubbart C, Hart L, Jeffery B, Drury E, Mead D, Kekre M, Campino S, Manske M, Cornelius VJ, MacInnis B, Rockett KA, Miles A, Rayner JC, Fairhurst RM, Nosten F, Price RN, Kwiatkowski DP. Genomic analysis of local variation and recent evolution in Plasmodium vivax
. Nat Genet 2016;48:959-964.
- 24. Hupalo DN, Luo Z, Melnikov A, Sutton PL, Rogov P, Escalante A, Vallejo AF, Herrera S, Arevalo-Herrera M, Fan Q, Wang Y, Cui L, Lucas CM, Durand S, Sanchez JF, Baldeviano GC, Lescano AG, Laman M, Barnadas C, Barry A, Mueller I, Kazura JW, Eapen A, Kanagaraj D, Valecha N, Ferreira MU, Roobsoong W, Nguitragool W, Sattabonkot J, Gamboa D, Kosek M, Vinetz JM, González-Cerón L, Birren BW, Neafsey DE, Carlton JM. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax
. Nat Genet 2016;48:953-958.
- 25. Zhong D, Bonizzoni M, Zhou G, Wang G, Chen B, Vardo-Zalik A, Cui L, Yan G, Zheng B. Genetic diversity of Plasmodium vivax malaria in China and Myanmar. Infect Genet Evol 2011;11:1419-1425.
- 26. Bastos MS, da Silva-Nunes M, Malafronte RS, Hoffmann EH, Wunderlich G, Moraes SL, Ferreira MU. Antigenic polymorphism and naturally acquired antibodies to Plasmodium vivax merozoite surface protein 1 in rural Amazonians. Clin Vaccine Immunol 2007;14:1249-1259.
- 27. Parobek CM, Bailey JA, Hathaway NJ, Socheat D, Rogers WO, Juliano JJ. Differing patterns of selection and geospatial genetic diversity within two leading Plasmodium vivax candidate vaccine antigens. PLoS Negl Trop Dis 2014;8:e2796.
- 28. Moore SJ, Min X, Hill N, Jones C, Zaixing Z, Cameron MM. Border malaria in China: knowledge and use of personal protection by minority populations and implications for malaria control: a questionnaire-based survey. BMC Public Health 2008;8:344.
- 29. Zhang HW, Liu Y, Zhang SS, Xu BL, Li WD, Tang JH, Zhou SS, Huang F. Preparation of malaria resurgence in China: case study of vivax malaria re-emergence and outbreak in Huang-Huai Plain in 2006. Adv Parasitol 2014;86:205-230.
- 30. Dias S, Longacre S, Escalante AA, Udagama-Randeniya PV. Genetic diversity and recombination at the C-terminal fragment of the merozoite surface protein-1 of Plasmodium vivax (PvMSP-1) in Sri Lanka. Infect Genet Evol 2011;11:145-156.
- 31. Thakur A, Alam MT, Sharma YD. Genetic diversity in the C-terminal 42 kDa region of merozoite surface protein-1 of Plasmodium vivax (PvMSP-1(42)) among Indian isolates. Acta Trop 2008;108:58-63.
- 32. Cui L, Yan G, Sattabongkot J, Cao Y, Chen B, Chen X, Fan Q, Fang Q, Jongwutiwes S, Parker D, Sirichaisinthop J, Kyaw MP, Su XZ, Yang H, Yang Z, Wang B, Xu J, Zheng B, Zhong D, Zhou G. Malaria in the Greater Mekong Subregion: heterogeneity and complexity. Acta Trop 2012;121:227-239.
- 33. Zhou XN, Bergquist R, Tanner M. Elimination of tropical disease through surveillance and response. Infect Dis Poverty 2013;2:1.
- 34. Chen SB, Ju C, Chen JH, Zheng B, Huang F, Xiao N, Zhou X, Ernest T, Zhou XN. Operational research needs toward malaria elimination in China. Adv Parasitol 2014;86:109-133.