Abstract
Development of an effective vaccine is critically needed for the prevention of malaria. One of the key antigens for malaria vaccines is the apical membrane antigen 1 (AMA-1) of the human malaria parasite Plasmodium falciparum, the surface protein for erythrocyte invasion of the parasite. The gene encoding AMA-1 has been sequenced from populations of P. falciparum worldwide, but the haplotype diversity of the gene in P. falciparum populations in the Greater Mekong Subregion (GMS), including Thailand, remains to be characterized. In the present study, the AMA-1 gene was PCR amplified and sequenced from the genomic DNA of 65 P. falciparum isolates from 5 endemic areas in Thailand. The nearly full-length 1,848 nucleotide sequence of AMA-1 was subjected to molecular analyses, including nucleotide sequence diversity, haplotype diversity and deduced amino acid sequence diversity and neutrality tests. Phylogenetic analysis and pairwise population differentiation (Fst indices) were performed to infer the population structure. The analyses identified 60 single nucleotide polymorphic loci, predominately located in domain I of AMA-1. A total of 31 unique AMA-1 haplotypes were identified, which included 11 novel ones. The phylogenetic tree of the AMA-1 haplotypes revealed multiple clades of AMA-1, each of which contained parasites of multiple geographical origins, consistent with the Fst indices indicating genetic homogeneity or gene flow among geographically distinct populations of P. falciparum in Thailand’s borders with Myanmar, Laos and Cambodia. In summary, the study revealed novel haplotypes and population structure needed for the further advancement of AMA-1-based malaria vaccines in the GMS.
-
Key words: Plasmodium falciparum, genetic polymorphism, human malaria, vaccine, antigen
INTRODUCTION
Malaria is a major tropical disease that is responsible for 0.4–0.5 million deaths worldwide [
1], with the vast majority of severe and fatal cases being confined to pregnant women and children of less than 5 year of age [
2,
3]. To reduce the burden of malaria, effective vaccines against the respective parasites, especially for
Plasmodium falciparum, are indispensable tools. Vaccines against the pathogenic blood stage of the malaria parasites would be useful not only in speeding up parasite clearance, but also in limiting the spread of anti-malarial drug resistance, which is now highly prevalent in Southeast Asia and many parts of the world [
4]. One of the major obstacles to vaccine development is the antigenic diversity of the genes in natural parasite populations, as demonstrated by whole genome sequencing [
5], which allows the parasites to escape host immune responses. The proteins of blood stage malaria parasites, such as merozoite surface proteins, have been identified as promising targets of immune responses, but they are reported to be highly polymorphic [
6]. Because a high degree of polymorphism occurs in field isolates, the polymorphism of the antigen-coding genes must be extensively studied before the development of an effective vaccine can be accomplished.
One of the leading candidate antigens for the development of a blood (erythrocyte) stage vaccine against
P. falciparum is the apical membrane antigen 1 (
AMA-1). The
AMA-1 protein is initially localized at the apical end of the merozoite that binds to the erythrocyte [
7]. In addition,
AMA-1 has been shown to be expressed in the sporozoite, which is the stage that participates in hepatocyte invasion [
8]. The full-length protein is comprised of 622 amino acid residues, has a molecular weight of 82 kDa, and can be divided into the 2 regions of a 550-amino acid extracellular region at the N-terminal and a transmembrane region at the C-terminal [
9]. The extracellular domain (ectodomain) is further divided into 3 domains (domains I, II, and III) and contains 16 Cys residues forming 8 intra-molecular disulfide bonds responsible for maintaining the 3D structure of proteins [
10]. The location of the disulfide bridges are highly conserved in all
Plasmodium species. Upon egression of the merozoite from an infected erythrocyte, the N-terminal region (prodomain) of the
AMA-1 protein is cleaved into a 66 kDa fragment, which is translocated to the neck of rhoptry through interaction with rhoptry neck protein 2 (RON2) [
11]. Subsequently, during the merozoite invasion of erythrocytes, secondary proteolytic cleavage occurs, resulting in the 48 kDa and 44 kDa fragments [
12]. Disruption of this proteolytic processing with antibodies against
AMA-1 has been shown to inhibit erythrocyte invasion [
13]. Furthermore, examination of the binding activity of
P. falciparum AMA-1-derived peptides revealed that the ectodomain of
AMA-1 possesses the erythrocyte binding activity and is a putative target of protective immune responses [
14]. The C-terminal portion of the protein is membrane-bound and carried into the invaded erythrocyte. Although the cytoplasmic tail of
AMA-1 is not required for the correct trafficking and surface translocation, it has been shown to be essential for
AMA-1 functions [
15].
Currently,
AMA-1 recombinant vaccines based on the
P. falciparum 3D7 and FVO strains are being tested in Phase 1 and 2 clinical studies, and the vaccines have shown promising results [
16–
20]. Nevertheless, one study using antibodies raised against recombinant
AMA-1 from
P. falciparum strain 3D7 (the genome reference strain, [
21]) revealed that the anti-
AMA-1 antibodies were potent inhibitors of 3D7 homologous strains but were less effective against strains with heterologous
AMA-1 haplotypes [
22]. This suggested that sequence variation within the
AMA-1 gene allows the parasites to escape the host immune responses, thereby posing a significant challenge for vaccine design. Accordingly, an understanding of the natural variation within the
AMA-1 gene sequence in the parasite populations will have an important practical implication for the design of future
AMA-1-based vaccines against
P. falciparum.
AMA-1 is encoded by a single-copy, intron-less gene that spans a 1,869 base pair (bp) region on chromosome 11 (PF11_0344) [
21]. As a vaccine candidate, the genetic diversity of the
AMA-1 gene has been a subject of intensive research. Detailed molecular studies on the polymorphism of the
P. falciparum AMA-1 gene have been conducted in various endemic areas worldwide, including the Middle East and South Asia [
23–
28], Oceania [
29–
31], Africa [
31–
39], and South America [
40–
41]. Whereas the majority of the
AMA-1 sequences spanned the regions of domain I or the ectodomain (domains I-III) of the gene, there are currently only limited datasets in which the full-length sequences of the
AMA-1 gene are reported [
31,
40,
42]. In addition, there is only limited
AMA-1 sequence data from
P. falciparum populations in Southeast Asia, including the Thailand-Myanmar border and none from the Thailand-Cambodia and Thailand-Laos borders, where the multidrug resistant parasites are often detected and the vaccines will be needed most [
43,
44]. Rather, within this region,
AMA-1 sequence data is mainly derived from small local parasite populations in Western Thailand [
31,
42,
45] and near the China-Myanmar border [
46,
47]. Furthermore, none of these studies have combined the data to generate a global view of the
AMA-1 diversity in
P. falciparum populations in Thailand.
To address this deficiency, this study aimed to determine the sequence diversity of the full-length sequences of AMA-1 in P. falciparum collected from 5 geographical locations near Thailand’s national borders. Comparative genetic analysis of the AMA-1 gene between P. falciparum isolates in Thailand and those of global geographical regions were performed. The outcome of this study will provide an insight into the nature and evolution of AMA-1 polymorphisms, which is a key feature for vaccine development.
MATERIALS AND METHODS
Origins of the P. falciparum malaria parasite isolates used in this study
Natural parasite isolates of
P. falciparum used in the present study were originally obtained from 68 patients in 5 malaria endemic localities in Thailand, including Mae Hong Son, Kanchaburi, Ranong, Trat and Ubon Ratchatani (
Supplementary Fig. 1). The parasite collections were conducted between 2001 and 2010. The blood stage malaria parasites were subjected to in vitro cultivation and maintained at the Department of Biology, Faculty of Science, Chulalongkorn University, Bangkok, Thailand. The species of the malaria parasites were confirmed by microscopic examination of thin blood films and by genotyping with genetic markers, including microsatellites [
48] and the merozoite surface protein-1 (MSP-1) and merozoite surface protein-3 (MSP-3) genes [
49,
50].
Merozoites of
P. falciparum were expanded in the laboratory using a in vitro culture protocol. Once the levels of parasitemia reached 5–10%, the parasites were harvested and used for genomic DNA extraction using a phenol/chloroform extraction method [
48–
50]. In brief, the parasitized erythrocytes were lysed in 0.05% (w/v) saponin in phosphate buffered saline pH 7.4 and then incubated in lysis buffer overnight. The cell lysates were treated sequentially with (i) saturated phenol, (ii) 25: 24: 1 (v/v/v) phenol: chloroform: isoamyl alcohol and (iii) chloroform prior to addition of 3 M sodium acetate and absolute ethanol for precipitation of the genomic DNA. The dried DNA pellets were dissolved in TE (10 mM Tris.HCl/1 mM EDTA, pH 8.0) buffer and stored at -20°C until use.
The PCR reaction for amplification of the
P. falciparum AMA-1 gene was prepared in a 50-μl volume containing 5–10 ng genomic DNA, 0.5 μmol of each of the For-AMA1 and Rev-AMA1 primers (
Supplementary Table S1), 0.5 mM MgCl
2 and 1 unit of Faststart
Taq DNA polymerase (Roche, Mannheim, Germany) in 1× Taq buffer. Thermal cycling was performed at 95°C for 10 min, followed by 40 cycles of 95°C for 1 min, 60°C for 1 min and 72°C for 2 min, and then a final 72°C for 10 min. The PCR products were analyzed by agarose gel electrophoresis. The PCR amplicons generated from the genomic DNA of 68 parasite isolates that produced a single PCR amplicon were then submitted to commercial DNA sequencing at Bioneer (Daejeon, Korea) using the primers shown in
Supplementary Table S1. DNA sequencing was performed bidirectionally using BigDye v3.1 and analyzed on an ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, California, USA). The obtained DNA sequences were aligned and contigs were assembled using Sequencher V. 4.5 (Gene Codes). The quality of DNA sequences and all SNPs were assessed by visual inspection to ensure that there were no gaps in the alignment.
Full-length (−1.8 kb) sequences of
AMA-1 (n=134 alleles) from
P. falciparum populations in Thailand were obtained from the nucleotide databases at the NCBI [
31,
32,
40,
42] (
Supplementary Table S2). The
AMA-1 sequences by Polley et al. [
45] were not included in the population genetic analysis because the only 1.3-kb sequences were available. Each unique sequence of
P. falciparum AMA-1 was treated as a unique haplotype, and the haplotype files were created using the DnaSP 5.0 software [
51]. Average number of pairwise nucleotide differences (
K), nucleotide diversity (π), number of haplotypes (H) and haplotype diversity (Hd) were calculated using MEGA software [
52]. The π estimated average number of substitutions between any 2 sequences, assuming that the sample is random. The distribution of the genetic diversity across the
AMA-1 gene was also plotted using a sliding window with a window length of 100 bp and moving the window in steps of 3 bp in the DnaSP 5.0 software to identify the region(s) of
AMA-1 that had accumulated polymorphisms. The π values were calculated in each window, and the values of π from each window were then plotted against the nucleotide position.
The mean number of non-synonymous substitutions per non-synonymous site (d
N) and synonymous mutations per synonymous site (d
S) within each isolate were estimated using Nei and Gojobori’s method [
53], with the Jukes and Cantor correction [
54], as implemented in the MEGA software. The significance of any differences between d
N and d
S were tested with the Z-test of selection (
P <0.05), where a d
N/d
S ratio of greater than 1 at the 95% confidence interval was taken as evidence of positive diversifying selection. In addition, 3 population genetic tests of neutrality (Tajima’s
D, and Fu and Li’s
D* and
F* tests) were used for detecting the signature of positive selection, that is whether polymorphism in
AMA-1 sequences occurred at higher or lower frequencies than expected under a neutral model [
55,
56]. Sliding window plots, with a window length of 100 bases and a step size of 3 bp, were also generated if there were specific regions with a significant departure from neutrality (
P <0.05).
Analysis of linkage disequilibrium was performed between nucleotide sites at which the frequency of the minor allele was >0.1 using an
R2 index with Fisher’s exact test of significance. The minimum number of recombination events in the history of samples was estimated according to the method of Hudson and Kaplan [
57]. All calculations for the neutrality tests, linkage disequilibrium and population recombination parameters were performed in the DnaSP 5.0 software.
The
P. falciparum AMA-1 sequences from natural isolates in Asia, Oceania, Africa and South America were downloaded from the NCBI database (
Supplementary Table S2). Nucleotide sequences corresponding to domain I from positions 445–906, corresponding to amino acid residues 149–302, were included in the population differentiation analysis. Arlequin suite version 3.5 software was used to calculate Wright’s fixation index (
Fst) to evaluate the differences in the distribution patterns (ratios) of
AMA-1 haplotypes between different geographic populations of
P. falciparum [
58]. Statistical significance was accepted at a
P-value of <0.05.
The analysis involved 47 nucleotide sequences (unique haplotypes) of
P. falciparum AMA-1 in Thailand and one
AMA-1 sequence from the chimpanzee malaria parasite
Plasmodium reichenowi (NCBI accession no. AJ252087; [
59]) that served as an out group. There were a total of 1,815 positions in the final dataset (from nt positions 25–1,838). The evolutionary history was inferred using the maximum likelihood (ML) method based on the general time reversible model. Initial tree(s) for the heuristic search were obtained automatically by applying neighbor-joining (NJ) and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum composite likelihood (MCL) approach, and then selecting the topology with a superior log likelihood value. The rate variation model allowed for some sites to be evolutionarily invariable ([+
I], 46.4463% sites). The evolutionary history was inferred based on the general time reversible with the Hasegawa-Kishino-Yano with gamma distribution shape parameter (HKY+G) model. Codon positions included were 1st+2nd+3rd+Noncoding. The reliability of the ML tree was accessed by the bootstrap method with 1,000 pseudo-replicates. Evolutionary analyses were conducted in MEGA 7.
RESULTS
Nucleotide sequence analysis of AMA-1
The PCR amplifications of AMA-1 from DNA templates of 68 P. falciparum isolates all yielded a single PCR amplicon with an estimated size of 1,800 bp. and were then sequenced. DNA sequences from 3 parasite isolates from Mae Hong Son province (MH07, MH09, and MH28) had ambiguous nucleotide sequences, likely due to the presence of multiple infections, and so were excluded from the analysis. Thus, the 1,848-bp nucleotide sequence fragment of the AMA-1 gene, corresponding to nucleotide positions 10–1,857 of P. falciparum 3D7, from 65 P. falciparum isolates in Thailand were analyzed.
Multiple sequence alignment of
AMA-1 identified 60 single nucleotide polymorphisms (SNPs). Of these, 58 and 2 SNPs resulted in non-synonymous and synonymous mutations, respectively. 4 sites (nucleotide positions 586, 590, 598, and 922) were trimorphic loci, while the remaining 56 sites were dimorphic loci. The 60 SNPs were distributed in 51 polymorphic codons (
Supplementary Table S3). Forty-three codons encoded dimorphic amino acids, 6 codons with 2 SNPs encoded trimorphic amino acids, while the other codon with 2 SNPs encoded 4 types of amino acids. In addition, there was one codon with 3 SNPs encoding 6 different amino acids. Although the majority of these SNPs have been reported in earlier studies [
31,
42,
45], an SNP at nucleotide position 730 (GAT, TAT) is reported here for the first time.
The SNPs were unevenly distributed across the
AMA-1 gene. As shown in
Supplementary Table S3, 34, 6, and 7 SNPs were located in domains I, II and III, respectively, whereas 2 and 1 SNPs were located in the flanking regions between domains I and II and between domains II and III, respectively. The other 10 SNPs were detected in the prosequence, the transmembrane domain, and the intracellular domain of
AMA-1. The
K-value for full-length
AMA-1 in samples of Thailand was 20.798 (
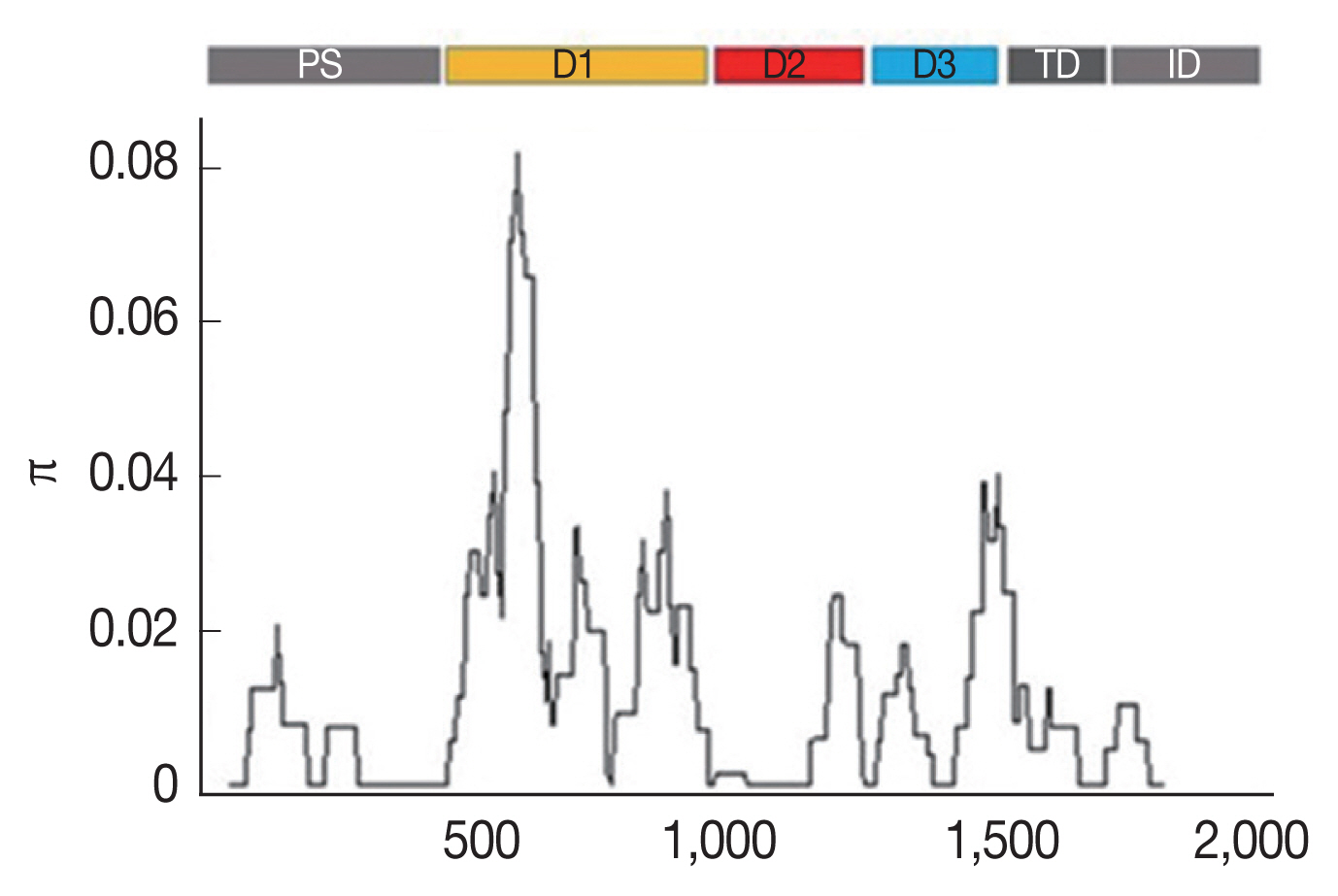
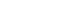
Table 1). The highest nucleotide difference was found at domain I (K=12.002), while the lowest was found at the transmembrane domain and intracellular domain (K=1.484). The analysis of the nucleotide sequence diversity (π) of domains I, II and III revealed π indices of 1.6449, 0.0058, and 0.1468, respectively, with an overall nucleotide sequence diversity (π) of
AMA-1 of 0.01141 (
Table 1;
Fig. 1). Together, these results indicated that the most polymorphic region of the gene was in domain I.
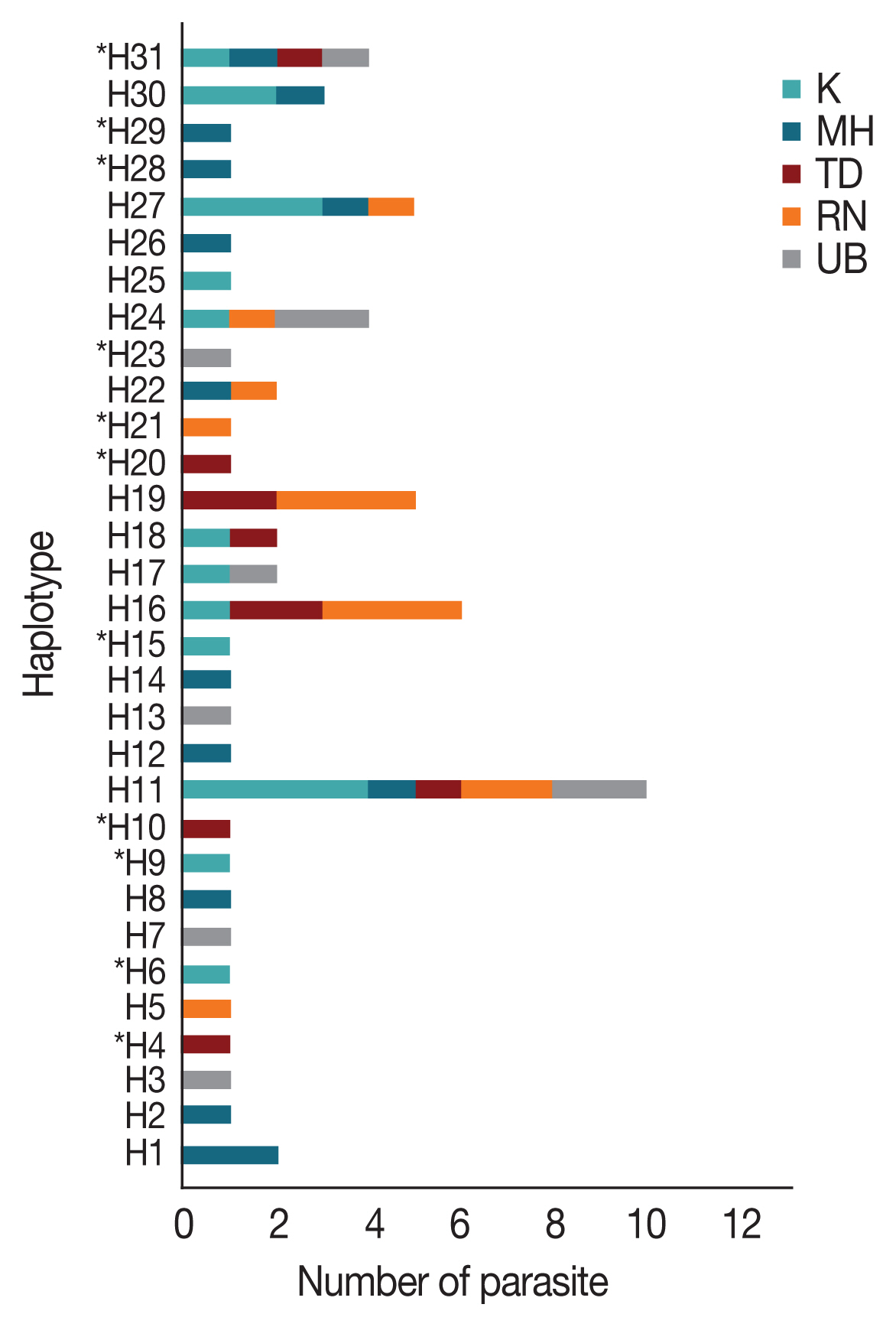
Multiple sequence alignment of the
AMA-1 gene of 65
P. falciparum isolates also identified 31 unique haplotypes (H) of the
AMA-1 sequences. The haplotype diversity index (Hd) of
AMA-1 was estimated to be 0.952. Twenty-one
AMA-1 haplotypes were detected in single localities. On the Thailand-Myanmar borders, 6, 6, and 2 haplotypes were detected in Mae Hong Son, Kanchanaburi and Ranong provinces, respectively. Additionally, 4 and 5 haplotypes were found only in Ubon Ratchatani and Trat, respectively, near the Thailand-Laos and Thailand-Cambodia borders, respectively. Of the 31 haplotypes identified, H11 was the most abundant and was distributed in all the sampled localities (
Fig. 2). The haplotype analysis also showed that 13 and 3 of these haplotypes had previously been reported in
P. falciparum populations in Thailand and Papua New Guinea, respectively [
31,
32,
40] (
Supplementary Table S4). The same
AMA-1 haplotypes were also detected in parasites from Myanmar and 11 parasite isolates originated from Western Thailand (e.g. Mae Hong Son, Kanchanaburi and Ranong) [
47]. In addition, 3 haplotypes were found to match the
AMA-1 sequences of
P. falciparum laboratory strains. Haplotypes H3 and H7 also matched the
AMA-1 sequences of parasite isolates Dd2 and D10, respectively, from Indochina [
32], while H19 corresponded to the sequence of the reference strain 3D7 [
21]. As a result, the present study has identified 11 novel
AMA-1 haplotypes that were unique to
P. falciparum populations in Thailand (
Fig. 2). When the
AMA-1 sequence data from the present study (n=65 alleles) were combined with the full-length
AMA-1 sequences from earlier studies (n=134 alleles) [
31,
32,
40,
42], a total of 47 unique haplotypes in 199
AMA-1 alleles were identified in the parasite populations in Thailand, with K, Hd and π values of 20.840, 0.9410, and 0.1128, respectively.
To identify the signature of natural selection in
AMA-1, 3 within-population tests for neutrality (Tajima’s
D, Fu and Li’s
D* and Fu and Li’s
F* tests) were performed using the 65
AMA-1 alleles in the present study. The Tajima’s
D and Fu and Li’s
F* values were significant, with values of 1.906 (
P <0.05) and 1.6448 (
P <0.05), respectively. However, although the Fu and Li’s
D* value (
D*=1.03967; >0.10) was positive it was not significant. When the
AMA-1 sequences (134 alleles) in Thailand from previous studies [
31,
32,
40,
42] were combined with the 65 alleles of this study and analyzed, similar results were obtained with significant Tajima’s
D and Fu and Li’s
F* values of 2.200 (<0.05) and 2.0362 (
P <0.05), respectively, while the Fu and Li’s
D* value (
D*=1.02317;
P >0.10) remained positive but insignificant. These results indicated a tendency for positive diversifying selection in the
AMA-1 gene. To further examine the possibility of natural selection, the d
N/d
S ratio was calculated using the sequences of the 65 alleles. The d
N/d
S ratio was 5.8211 (greater than 1). Taken together, the results of the neutrality tests and the d
N/d
S ratio supported the view that the positive (diversifying) selection may contribute to the diversity of the
AMA-1 gene.
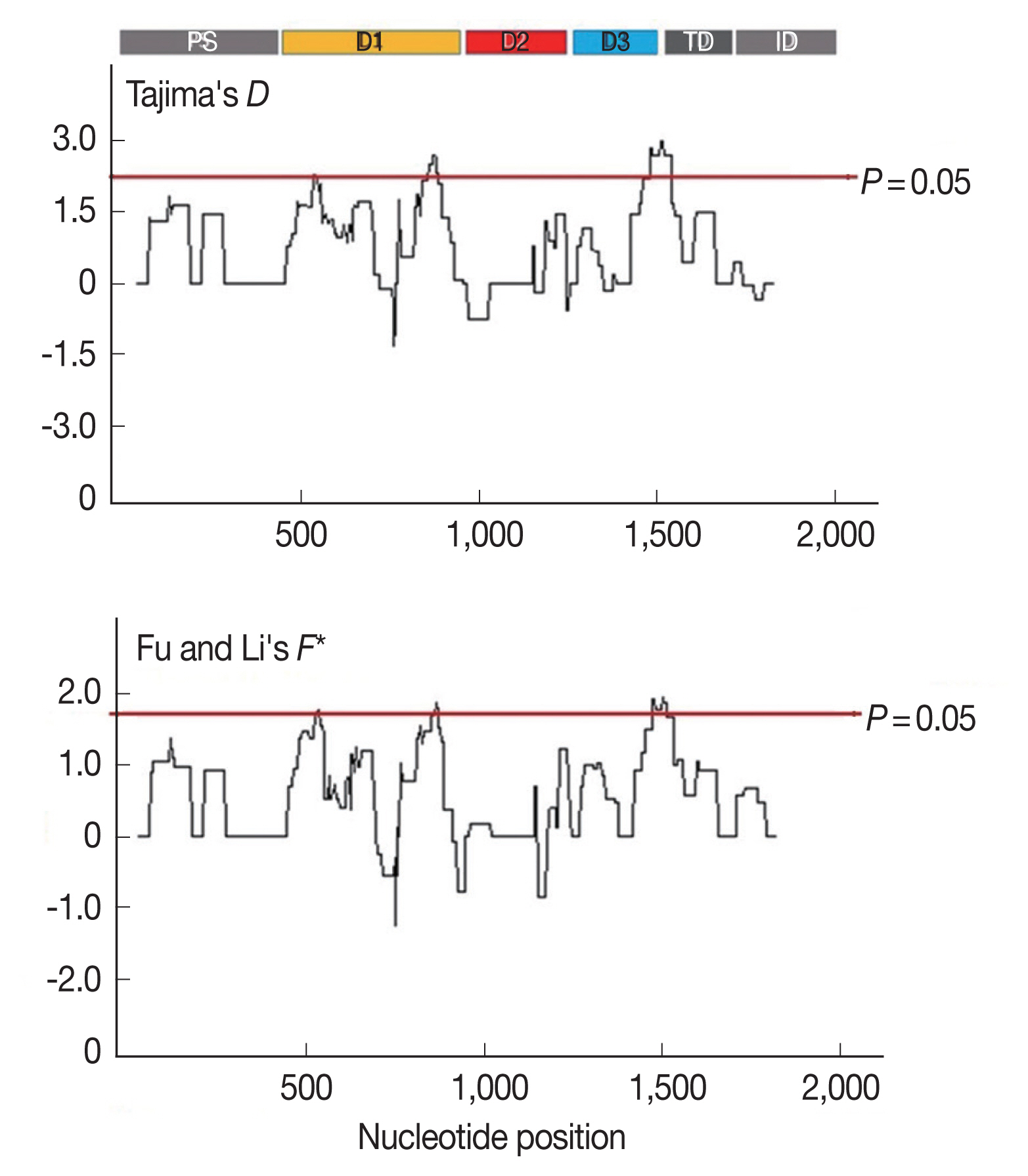
Furthermore, to determine whether specific regions of
AMA-1 were under positive selection, a sliding window plot analysis was performed (
Fig. 3). The values of
D and
F* were calculated based on a window of 100 bp moving in 3-bp steps. According to the Tajima’s
D test, 3 regions of
AMA-1 were found to be under positive selection: nucleotide positions 502–573 and 829–906 in domain I and 1,450–1,545 in domain III. Similar regions spanning nucleotide positions 502–585, 808–921, and 1,429–1,506 showed a significant departure from neutrality in the Fu and Li’s
F* test. Thus, it is likely that positive diversifying selection mainly contributed to the genetic polymorphism in domains I and III of
AMA-1.
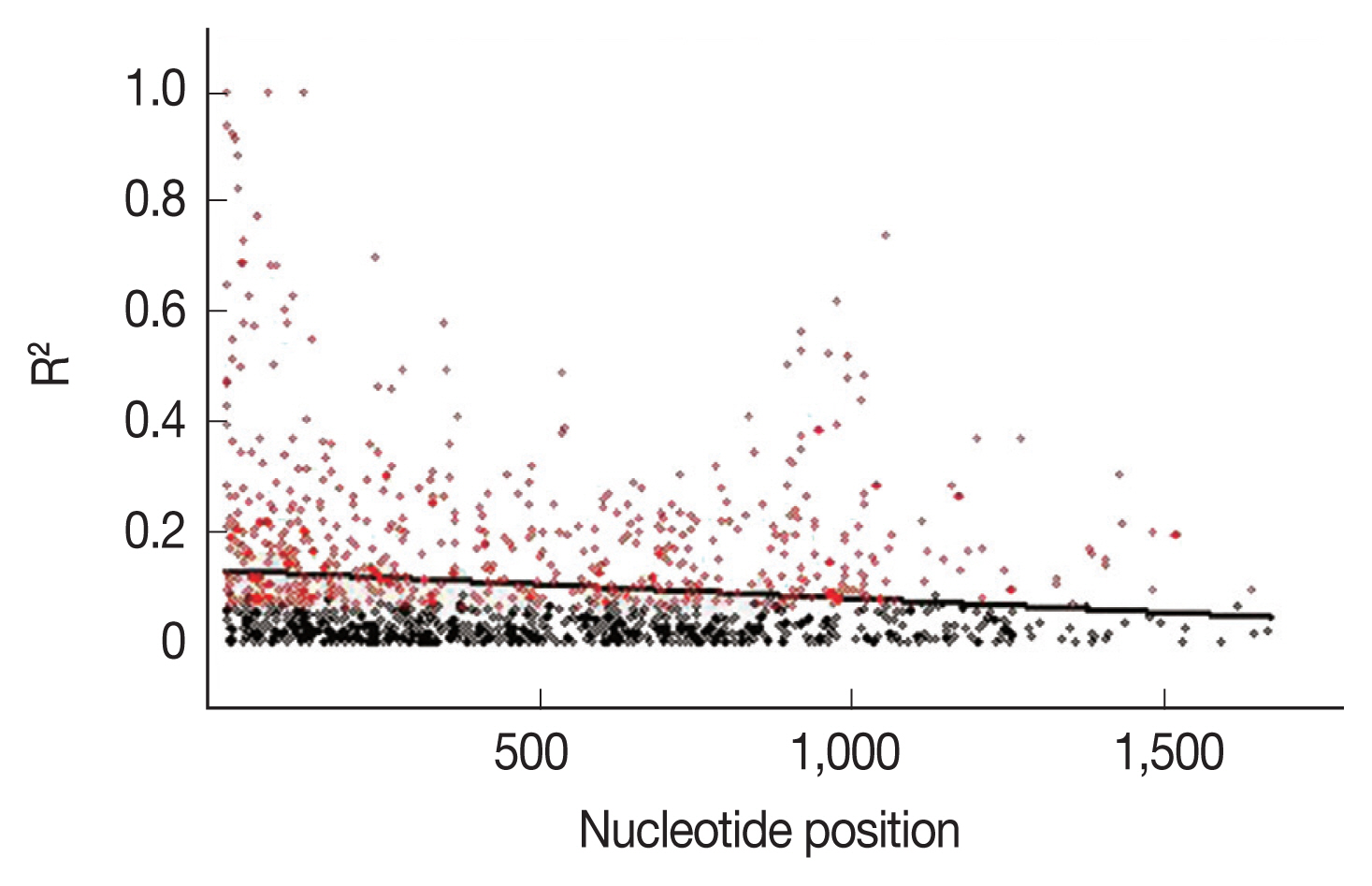
To further explain the high mutation rates at the
AMA-1 locus, linkage disequilibrium was calculated for all the non-singleton polymorphic residues and plotted against the nucleotide distance.
Fig. 4 demonstrated that the linkage disequilibrium index (
R2) declined rapidly with increasing nucleotide distance. It can be assumed that a high meiotic recombination rate may contribute to the increased polymorphism in the
AMA-1 locus. A minimum of 20 recombination events (Rm) was predicted to have occurred to give rise to the 31 haplotypes observed in these 65 alleles. The recombination parameter had high values of 0.0371 between adjacent nucleotide sites (R
a) and 68.6 for the whole sequence (R
b) in
P. falciparum populations in Thailand. Overall, this result was consistent with the previous analysis of recombinant parameters using the partial
AMA-1 sequences from
P. falciparum populations from Western Thailand [
45].
The distribution of the 65
AMA-1 alleles in
P. falciparum in Thailand (this study) also revealed different levels of genetic diversity in the different geographical populations. The highest genetic diversity of
AMA-1 was detected in
P. falciparum populations near the Thailand-Myanmar borders, with a total of 12 and 11 haplotypes being identified in Kanchanaburi and Mae Hong Son, respectively. On the other hand, the parasite population in Ubon Ratchatani had the lowest genetic diversity of
AMA-1, with a total of 8 haplotypes being identified. To compare the patterns of distribution of
AMA-1 haplotypes between
P. falciparum populations in Thailand, pairwise inter-population comparisons were performed for each population using Wright’s Fixation index (
Fst).
Table 2 shows that a significant
Fst value (
P =0.03) was found between the parasite populations in Mae Hong Son and Ranong, indicating potential population sub-division between these 2
P. falciparum populations. In contrast, the
Fst values from the other pairs of all 5 parasite populations were low and non-significant (
P >0.05). When the previously reported
AMA-1 haplotypes of
P. falciparum in Tak, Western Thailand [
31,
42,
45] were included in the
Fst analysis, the
Fst values remained low and non-significant between
P. falciparum in Tak and the other populations in Thailand. Therefore, this result inferred that the
P. falciparum parasite populations (or at least the
AMA-1 gene) circulating in this region were genetically homogeneous.
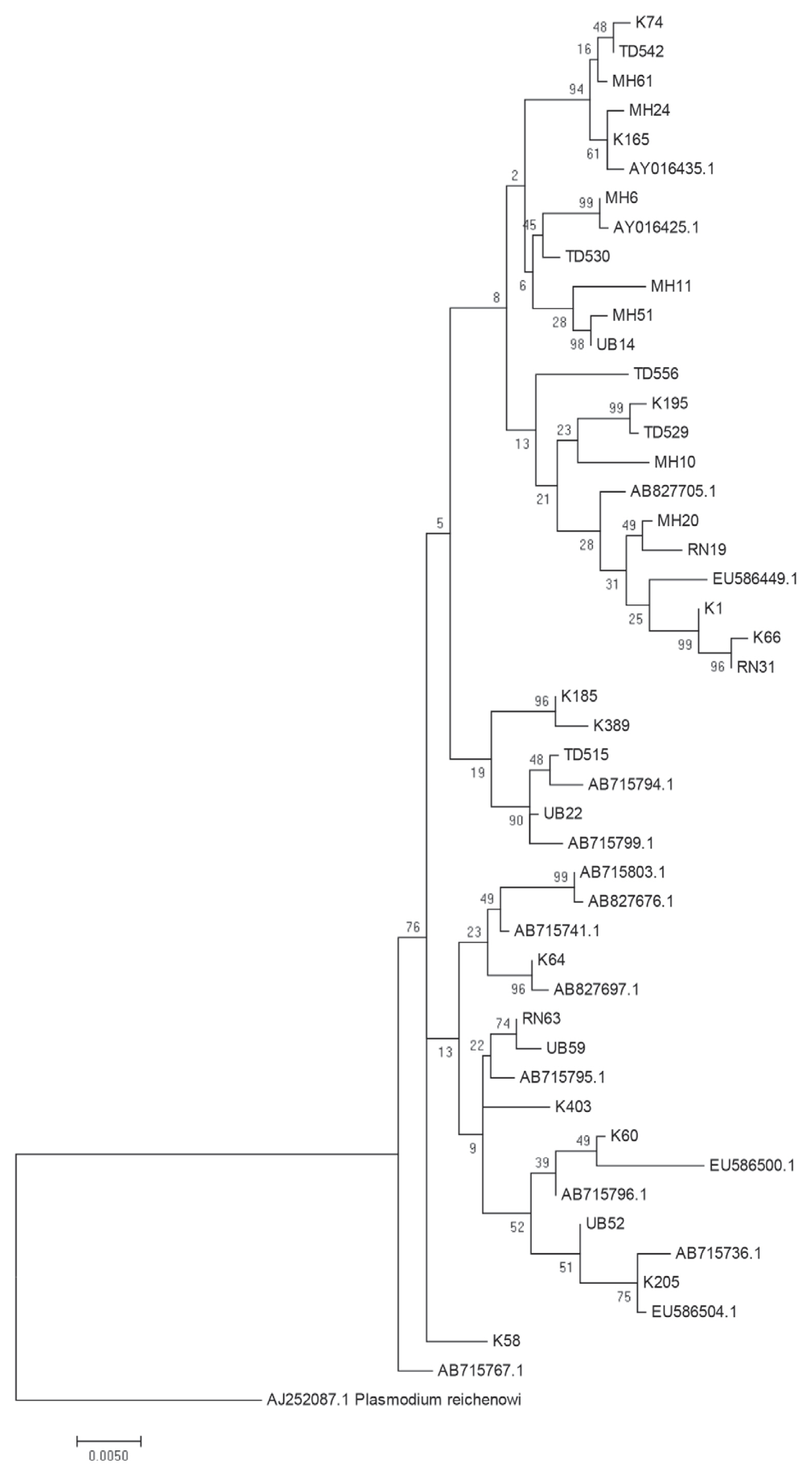
The above data provide suggestive evidence of the genetic flow between allopatric
P. falciparum populations near the borders of Thailand with Myanmar, Laos and Cambodia. To further examine the close genetic relationship of the
P. falciparum AMA-1 gene, ML-based phylogenetic trees of the 47 unique
AMA-1 haplotypes were constructed, with the highest log likelihood tree shown in
Fig. 5. The
AMA-1 haplotypes formed 3 major clades, where the
AMA-1 genes in each clade were from multiple geographical origins, reinforcing the idea that genetic recombination is very successful between closely related parasite populations.
Furthermore, sequences of
AMA-1 domain I from
P. falciparum populations in 16 countries worldwide, including 3 countries in each of Southeast Asia (Myanmar, the Philippines, and Malaysia), the Middle East and South Asia (India, Saudi Arabia, and Iran), and Oceania (The Solomon Islands, Vanuatu, and Papua New Guinea), 6 countries in Africa (The Gambia, Ghana, Tanzania, Nigeria, Kenya, and Mali) and one country in South America (Venezuela), were retrieved through a nucleotide database search at the NCBI website (
Supplementary Table S2). A total of 3,311 alleles, including the 65 Thai sequences from the present study, were obtained. Pairwise inter-population comparisons were performed between the
AMA-1 haplotypes of
P. falciparum in Thailand and those of the other countries. The
Fst values were significant between the
P. falciparum population in Thailand and other populations, except for Myanmar (
Table 2), suggesting a similar allelic distribution of the
AMA-1 gene in the Greater Mekong Subregion (GMS). The analysis also inferred that population differentiation has occurred between
P. falciparum in the GMS and other countries.
DISCUSSION
The present study aimed to investigate the extent of haplotype diversity in the
P. falciparum AMA-1 gene in Thailand. The main finding was that the polymorphic
P. falciparum AMA-1 gene within this region had as many as 31 haplotypes, which were characterized by 60 SNP loci across the gene. The SNPs were distributed mainly in the ectodomain of the
AMA-1 gene, which is in agreement with previous reports that domain I of
AMA-1 was the most polymorphic region [
36,
40,
45]. Nearly 2/5 (41%) of the
AMA-1 haplotypes identified in the present study had previously been detected in Tak, Western Thailand [
31,
32,
40], while the level of nucleotide diversity of
AMA-1 in
P. falciparum in Thailand was similar to a previous report on
P. falciparum populations in Myanmar [
46]. Interestingly, 5 haplotypes (16%) of the
AMA-1 gene identified in the present study were identical to those identified in
P. falciparum in Myanmar [
47], indicating the close genetic relatedness between
P. falciparum populations in Thailand and China-Myanmar. This hypothesis was supported by Wright’s statistics, which suggests that gene flow operates among
P. falciparum populations in the GMS. However, note that the
P. falciparum samples from Southern China and Vietnam were not included in this study and should be included in further studies to generate a more complete view of the
P. falciparum AMA-1 genetic diversity in Southeast Asia.
The Wright
Fst analysis also indicated that the population structure of
P. falciparum in Southeast Asia, based on the
AMA-1 gene, was significantly different from the populations in the Middle East, South Asia, Oceania, Africa and South America. This result was similar to a separate study using the
P. falciparum merozoite surface protein-3 gene [
49,
50]. The population subdivision among Asia, Africa and South America has also been reported using whole genome sequence data [
5]. The different population structures, using either the
AMA-1 gene or the whole genome sequences, reflect the different intensity and prevalence of malaria parasites in each geographical region. In Southeast Asian countries, the malaria transmission rate is low, seasonal and stable, while in Africa, malaria is holoendemic [
43,
47,
60]. Despite the different population structures, the number of
AMA-1 sequences shared among the parasite populations, however, suggests that similar selective forces act in the different geographical regions.
In addition to the population structure of the malaria parasite, sequence analysis of
AMA-1 alleles also revealed that amino acid substitution is generally favored with a clear signature of positive selection in the
AMA-1 locus among Thai parasite isolates. That is the number of non-synonymous substitutions per non-synonymous site significantly exceeded the number of synonymous substitutions per synonymous site [
61]. This adaptive evolution is presumably due to host immune selection. Additionally, the results from neutrality tests further supported positive diversifying selection operating at the
P. falciparum AMA-1 locus, while the window analysis of Tajima’s
D and Fu and Li’s
D* tests suggested that positive selection acts specifically on domains I and III. Although the values of Fu and Li’s
F* value test were not significant across the entire region, the positive values indicated the tendency of departure from neutral evolution and towards positive diversifying selection. This result concurs with the finding from previous studies in
P. falciparum in Nigerian and Thai populations, which showed that excessive polymorphism in domains I and III of
AMA-1 was maintained by positive selection, while no evidence of selection was detected for domain II [
33,
36,
45]. In addition, there is evidence that the polymorphic sites in domains I and III are targets of inhibitory antibodies [
62–
64]. Taken together, these results imply that domains I and III are potential targets for protective immunity and could be used as vaccine subunits.
Current
AMA-1 vaccines that are being tested in Phase I and II clinical trials are based on the sequence of
P. falciparum strain 3D7 and/or FVO [
19,
22,
65]. In the present study, only one (H19) of the 31
AMA-1 haplotypes matched the 3D7 strain. A previous analysis of the 135
AMA-1 alleles from
P. falciparum in Myanmar showed that none of them was identical to the 3D7 strain, although a few isolates had the
AMA-1 sequence of the FVO strain [
46]. Accordingly, it is postulated that monovalent or bivalent vaccines may offer little protection to the immunized subjects in the GMS. It has been shown using the rodent malaria model that the
AMA-1 vaccine elicited immune protection against parasites with the homologous
AMA-1 allele but not those with a heterologous allele [
16]. Likewise, the results from Phase II clinical trials showed that vaccinations with a monovalent
AMA-1 vaccine (FMP2.1/AS) in children in Mali conferred 20% efficacy that was strain-specific [
66]. Nonetheless, the analysis of global
AMA-1 haplotypes showed that several major alleles of
AMA-1 were shared between endemic areas regardless of geographical location [
32]. The inclusion of multiple
P. falciparum AMA-1 alleles as vaccines according to the local population structure of
P. falciparum may give a better protective efficacy of
AMA-1 if it is allele-specific [
67]. Hence, there is no doubt that estimates of molecular diversity of the major vaccine candidate,
AMA-1, have practical implications for the rational design of effective
AMA-1-based vaccines.
In conclusion, we have formed an initial AMA-1 sequence database of P. falciparum in Thailand comprised of baseline data of P. falciparum AMA-1 polymorphisms in natural isolates that originated from the national borders near Myanmar, Laos and Cambodia. Novel haplotypes of AMA-1 were found and strongly support the presence of positive selection acting on the gene. Because countries in Southeast Asia are entering the age of ASEAN Economic Community (AEC), with an increased movement of people between the countries, the continuous introduction and exposure of imported malaria cases is expected to increase. This would potentially contribute to an increased level of genetic recombination and polymorphism of the malaria parasite within and between populations in the AEC region, presenting new challenges to malaria elimination programs. An understanding of the genetic polymorphism in any vaccine candidate, including AMA-1, is necessary for malaria vaccine development in light of the extensive antigenic diversity and allele-specific immunity.
Notes
-
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Supplementary materials
Supplementary Fig. S1.
Plasmodium falciparum collection sites in Thailand. Mae Hong Son (MH), Kanchanaburi (K) and Ranong (RN) are situated in Western Thailand and share a border with Myanmar. Trat (TD) is located in Eastern Thailand and shares a border with Cambodia, while Ubon Ratchatani (UB) is in Northeastern Thailand and shares a border with Laos. n, the number of parasites. In total, 68 parasites isolates were analyzed in the present study.
Supplemenatary Table S1.
Nucleotide sequences of primers for amplification of P. falciparum AMA-1 gene and for DNA sequencing
Supplemenatary Table S2.
Sequences of the AMA-1 gene of P. falciparum populations in Asia, Oceania, Africa, and South America
Supplemenatary Table S3.
Polymorphic nucleotides in the AMA-1 gene of P. falciparum in Thailand
Supplemenatary Table S4.
NCBI accession numbers of the 31 AMA-1 haplotypes from P. falciparum in Thailand reported in the present study
ACKNOWLEDGMENTS
This work was supported by a research grant for the Development and Promotion of Science and Technology Talents Project (DPST) graduate with First Placement, Royal Thai Government to SP; and a grant from the Faculty of Science, Chulalongkorn University to LL. MK and SP were supported in part by a Special Task Force for Activating Research grant of Chulalongkorn University (GSTAR 6000731004-1).We thank Dr. Robert Butcher (Division of Research Affairs, Chulalongkorn University, Thailand) for language editing and proofreading.
Fig. 1Sliding window plots of the average pairwise nucleotide diversity (π) of the
AMA-1 gene of
P. falciparum in Thailand. The nucleotide diversity was plotted by a sliding window of 100 bp and a step size of 3 bp. Nucleotide positions 10–1,857 were included in the analysis. The positions of
AMA-1 sequences are numbered after
P. falciparum strain 3D7. Domains in
AMA-1 are subdivided into prosequence (PS), domain I (D1), domain II (D2), domain III (D3), transmembrane domain (TD) and intracellular domain (ID) [
12].
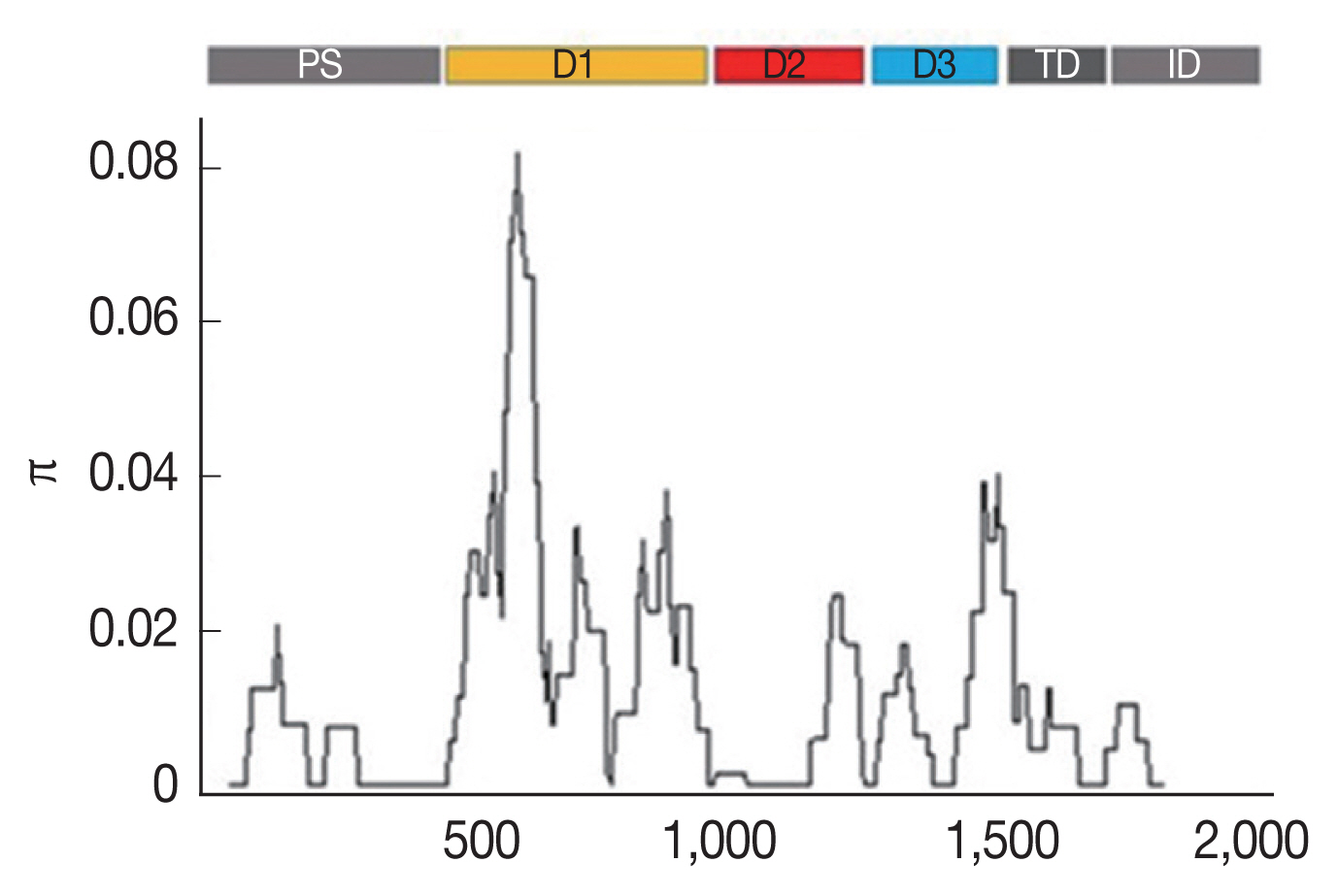
Fig. 2Distribution and frequency of the 31 AMA-1 haplotypes in Thailand. Asterisks (*) indicate the 11 novel haplotypes. H, haplotype; K, Kanchanaburi; MH, Mae Hong Son; RN, Ranong; TD, Trat; UB, Ubon Ratchatani.
Fig. 3Sliding window plots of Tajima’s D and Fu and Li’s F* statistics for the P. falciparum AMA-1 gene in Thai isolates. Nucleotide positions are according to the 3D7 sequence. Sliding window (100 bp) plots of all neutrality tests were performed at a step size of 3 bp. A brown line indicates a region where significant departure from neutrality occured (P<0.05). Significant D-values were detected between nucleotide positions 502–573, 829–906, and 1,450–1,545, while significant F* values were detected between nucleotide positions 502–585, 808–921, and 1,429–1,506.
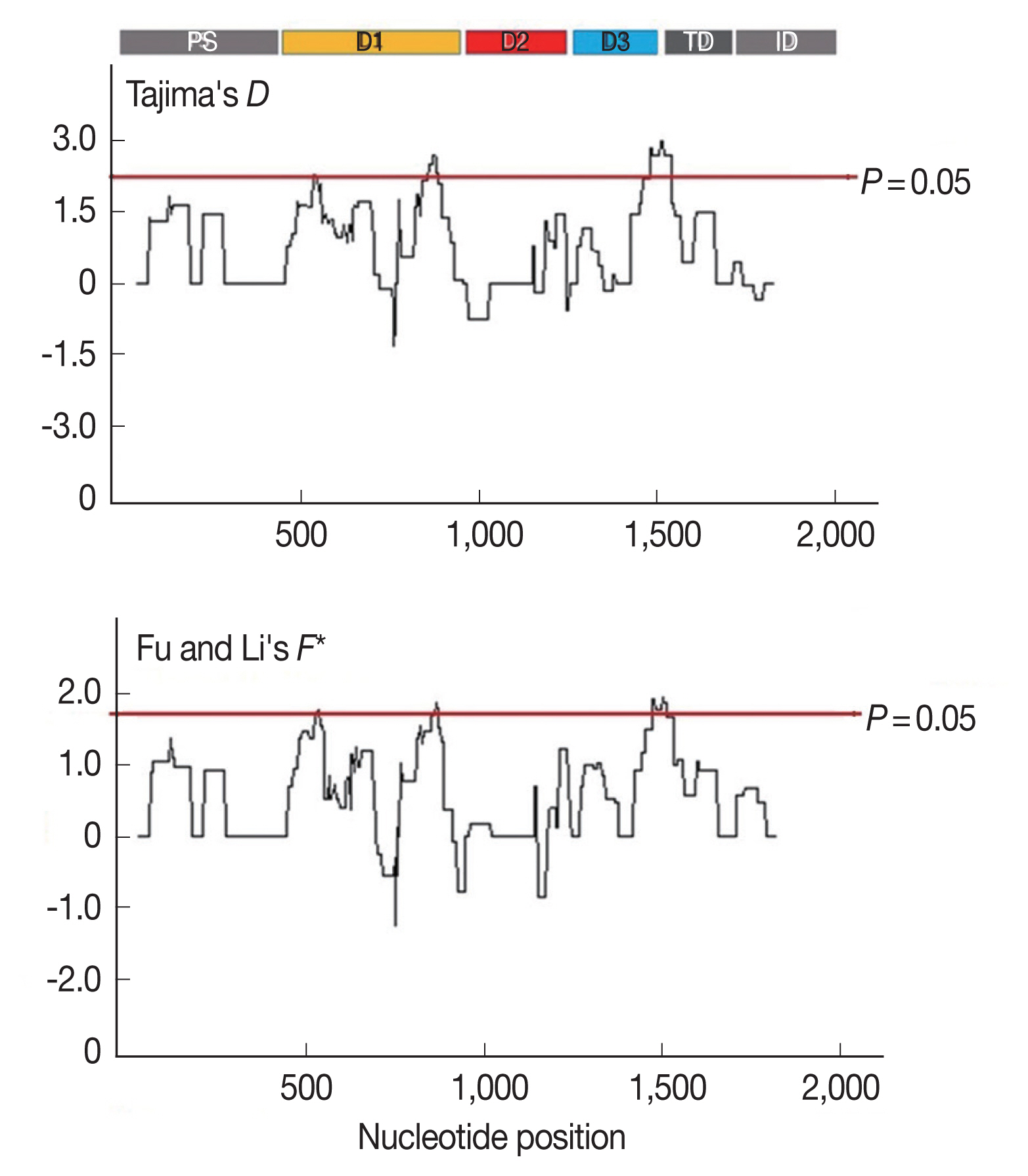
Fig. 4Linkage disequilibrium (LD) in P. falciparum populations in Thailand. Red dots indicate significant linkage disequilibrium (R2), as calculated by the Fisher’s exact test, while all others are shown as black circles. The regression line is represented by the trace line.
Fig. 5Maximum likelihood phylogenetic tree of the 47 unique haplotypes of the AMA-1 gene of P. falciparum in Thailand. The tree was constructed using the MEGA program from AMA-1 nucleotide sequences. The tree with the highest log likelihood (-4,803.3701) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Sequences are identified with their names of parasite isolates. The letters indicate the origin of isolates MH, Mae Hong Son; K, Kanchanaburi; RN, Ranong; TD, Trat; UB, Ubon Ratchatani. The tree was rooted using the sequence of the P. reichenowi AMA-1 gene as an out group. Bootstrap values of >50% are shown.
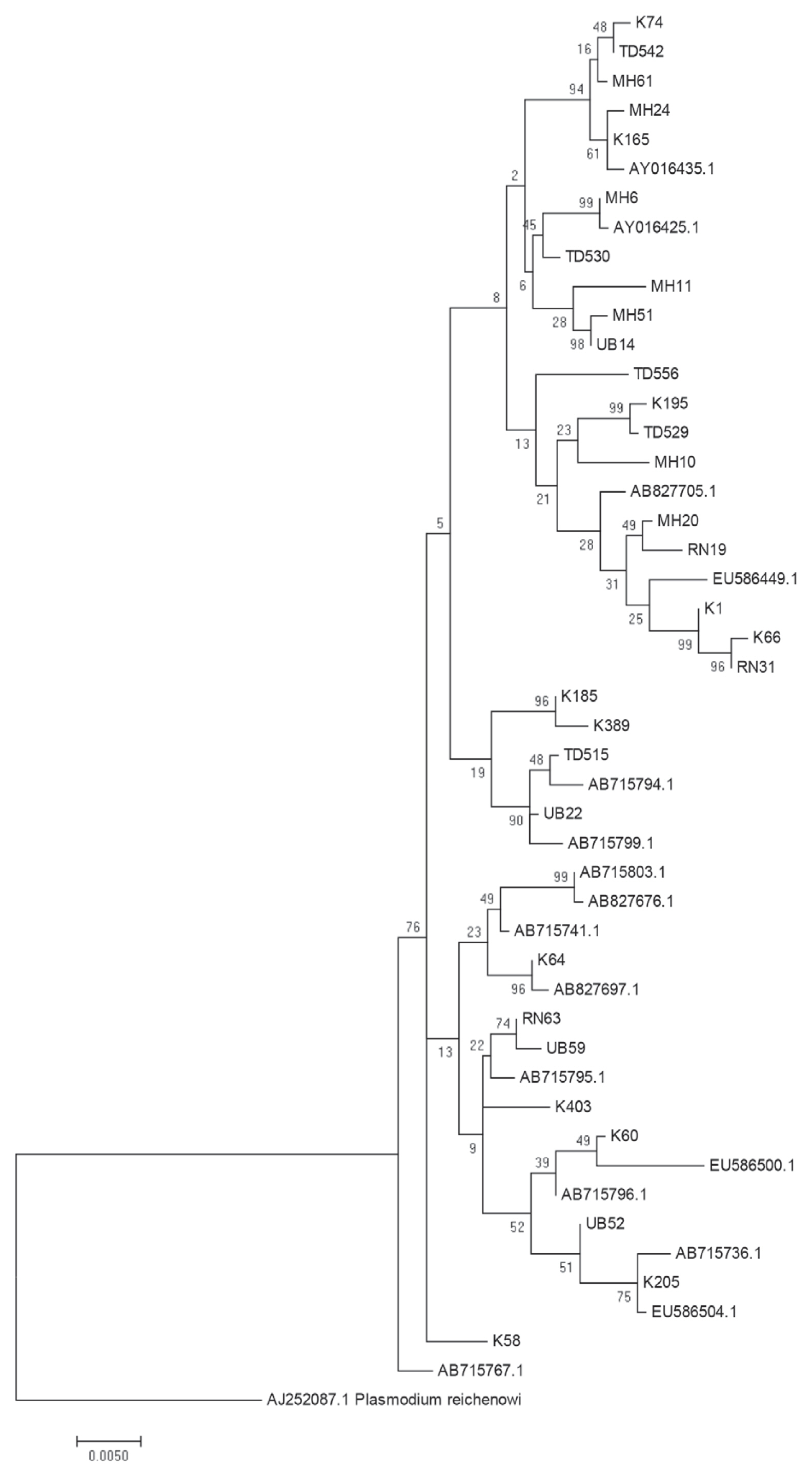
Table 1Pairwise Fst indices of AMA-1 alleles in P. falciparum in Thailand
Table 1
|
MH |
K |
RN |
TD |
UB |
|
K |
0.02044 |
|
|
|
|
|
P=0.29 |
|
|
|
|
|
|
RN |
0.08342 |
0.00108 |
|
|
|
|
P<0.05 |
P=0.42 |
|
|
|
|
|
TD |
0.02977 |
−0.00567 |
−0.04477 |
|
|
|
P=0.21 |
P=0.58 |
P=0.86 |
|
|
|
|
UB |
0.06928 |
−0.01413 |
0.01282 |
0.00371 |
|
|
P=0.07 |
P=0.63 |
P=0.36 |
P=0.45 |
|
|
|
Taka
|
0.01849 |
0.00095 |
−0.00064 |
0.00193 |
0.03024 |
|
P=0.17 |
P=0.40 |
P=0.42 |
P=0.43 |
P=0.10 |
Table 2Pairwise Fst indices of AMA-1 alleles in P. falciparum from 17 geographical populations
Table 2
|
Southeast Asia |
Middle East and South Asia |
Oceania |
SA |
Africa |
|
|
|
|
|
|
TH |
MM |
MY |
PH |
IN |
SA |
IR |
VU |
SB |
PG |
VE |
GM |
GH |
TZ |
NG |
KE |
ML |
|
MM |
−0.002 |
- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
P=0.35 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
MY |
0.007 |
0.009 |
- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
P<0.01 |
P<0.01 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
PH |
0.035 |
0.053 |
0.010 |
- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
P<0.01 |
P<0.01 |
P<0.01 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
IN |
0.054 |
0.051 |
0.130 |
0.111 |
- |
|
|
|
|
|
|
|
|
|
|
|
|
|
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SA |
0.060 |
0.065 |
0.033 |
0.030 |
0.050 |
- |
|
|
|
|
|
|
|
|
|
|
|
|
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
IR |
0.099 |
0.101 |
0.141 |
0.139 |
0.122 |
0.117 |
- |
|
|
|
|
|
|
|
|
|
|
|
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
|
|
|
|
|
|
|
|
|
|
|
|
|
VN |
0.115 |
0.120 |
0.086 |
0.085 |
0.114 |
0.069 |
0.177 |
- |
|
|
|
|
|
|
|
|
|
|
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
|
|
|
|
|
|
|
|
|
|
|
|
SB |
0.070 |
0.071 |
0.011 |
0.031 |
0.054 |
0.043 |
0.129 |
0.023 |
- |
|
|
|
|
|
|
|
|
|
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P =0.08 |
|
|
|
|
|
|
|
|
|
|
|
PG |
0.055 |
0.051 |
0.042 |
0.032 |
0.069 |
0.029 |
0.080 |
0.076 |
0.021 |
- |
|
|
|
|
|
|
|
|
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
|
|
|
|
|
|
|
|
|
|
VE |
0.251 |
0.231 |
0.306 |
0.158 |
0.199 |
0.214 |
0.171 |
0.169 |
0.222 |
0.103 |
- |
|
|
|
|
|
|
|
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
|
|
|
|
|
|
|
|
|
GM |
0.027 |
0.038 |
0.044 |
0.043 |
0.056 |
0.066 |
0.097 |
0.076 |
0.021 |
0.011 |
0.195 |
- |
|
|
|
|
|
|
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
|
|
|
|
|
|
|
|
GH |
0.035 |
0.035 |
0.063 |
0.047 |
0.051 |
0.022 |
0.087 |
0.100 |
0.050 |
0.015 |
0.201 |
0.002 |
- |
|
|
|
|
|
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P =0.12 |
|
|
|
|
|
|
|
TZ |
0.042 |
0.024 |
0.083 |
0.038 |
0.045 |
0.045 |
0.104 |
0.097 |
0.044 |
0.012 |
0.232 |
0.004 |
−0.002 |
- |
|
|
|
|
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P =0.26 |
P =0.22 |
|
|
|
|
|
|
NG |
0.041 |
0.076 |
0.099 |
0.029 |
0.066 |
0.047 |
0.063 |
0.115 |
0.041 |
0.011 |
0.191 |
−0.001 |
−0.006 |
0.003 |
- |
|
|
|
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P =0.06 |
P =0.23 |
P =0.10 |
|
|
|
|
|
KE |
0.037 |
0.088 |
0.050 |
0.040 |
0.054 |
0.033 |
0.099 |
0.108 |
0.055 |
0.020 |
0.203 |
0.005 |
0.001 |
−0.002 |
0.001 |
- |
|
|
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P =0.06 |
P =0.08 |
P =0.06 |
P =0.18 |
|
|
|
|
ML |
0.040 |
0.038 |
0.013 |
0.044 |
0.059 |
0.026 |
0.056 |
0.087 |
0.050 |
0.020 |
0.176 |
0.010 |
0.002 |
0.009 |
0.011 |
0.009 |
- |
|
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P<0.01 |
P =0.11 |
P<0.01 |
P <0.01 |
P<0.01 |
|
References
- 1. World Heath Organization. World Malaria Report, 2016. Geneva, Switzerland. World Heath Organization; 2016.
- 2. Fried M, Duffy PE. Malaria during Pregnancy. Cold Spring Harb Perspect Med 2017;7:a025551.
- 3. Maitland K. Severe malaria in African children - the need for continuing investment. N Engl J Med 2016;375:2416-2417.
- 4. Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han KT, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, Rahman MR, Hasan MM, Islam A, Miotto O, Amato R, MacInnis B, Stalker J, Kwiatkowski DP, Bozdech Z, Jeeyapant A, Cheah PY, Sakulthaew T, Chalk J, Intharabut B, Silamut K, Lee SJ, Vihokhern B, Kunasol C, Imwong M, Tarning J, Taylor WJ, Yeung S, Woodrow CJ, Flegg JA, Das D, Smith J, Venkatesan M, Plowe CV, Stepniewska K, Guerin PJ, Dondorp AM, Day NP, White NJ. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2014;371:411-423.
- 5. Manske M, Miotto O, Campino S, Auburn S, Almagro-Garcia J, Maslen G, O’Brien J, Djimde A, Doumbo O, Zongo I, Ouedraogo JB, Michon P, Mueller I, Siba P, Nzila A, Borrmann S, Kiara SM, Marsh K, Jiang H, Su XZ, Amaratunga C, Fairhurst R, Socheat D, Nosten F, Imwong M, White NJ, Sanders M, Anastasi E, Alcock D, Drury E, Oyola S, Quail MA, Turner DJ, Ruano-Rubio V, Jyothi D, Amenga-Etego L, Hubbart C, Jeffreys A, Rowlands K, Sutherland C, Roper C, Mangano V, Modiano D, Tan JC, Ferdig MT, Amambua-Ngwa A, Conway DJ, Takala-Harrison S, Plowe CV, Rayner JC, Rockett KA, Clark TG, Newbold CI, Berriman M, MacInnis B, Kwiatkowski DP. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature 2012;487:375-379.
- 6. Mu J, Awadalla P, Duan J, McGee KM, Keebler J, Seydel K, McVean GA, Su XZ. Genome-wide variation and identification of vaccine targets in the Plasmodium falciparum genome. Nat Genet 2007;39:126-130.
- 7. Triglia T, Healer J, Caruana SR, Hodder AN, Anders RF, Crabb BS, Cowman AF. Apical membrane antigen 1 plays a central role in erythrocyte invasion by Plasmodium species. Mol Microbiol 2000;38:706-718.
- 8. Silvie O, Franetich JF, Charrin S, Mueller MS, Siau A, Bodescot M, Rubinstein E, Hannoun L, Charoenvit Y, Kocken CH, Thomas AW, Van Gemert GJ, Sauerwein RW, Blackman MJ, Anders RF, Pluschke G, Mazier D. A role for apical membrane antigen 1 during invasion of hepatocytes by Plasmodium falciparum sporozoites. J Biol Chem 2004;279:9490-9496.
- 9. Marshall VM, Zhang L, Anders RF, Coppel RL. Diversity of the vaccine candidate AMA-1 of Plasmodium falciparum
. Mol Biochem Parasitol 1996;77:109-113.
- 10. Hodder AN, Crewther PE, Matthew ML, Reid GE, Moritz RL, Simpson RJ, Anders RF. The disulfide bond structure of Plasmodium apical membrane antigen-1. J Biol Chem 1996;271:29446-29452.
- 11. Srinivasan P, Beatty WL, Diouf A, Herrera R, Ambroggio X, Moch JK, Tyler JS, Narum DL, Pierce SK, Boothroyd JC, Haynes JD, Miller LH. Binding of Plasmodium merozoite proteins RON2 and AMA1 triggers commitment to invasion. Proc Natl Acad Sci USA 2011;108:13275-13280.
- 12. Howell SA, Withers-Martinez C, Kocken CH, Thomas AW, Blackman MJ. Proteolytic processing and primary structure of Plasmodium falciparum apical membrane antigen-1. J Biol Chem 2001;276:31311-31320.
- 13. Dutta S, Haynes JD, Moch JK, Barbosa A, Lanar DE. Invasion-inhibitory antibodies inhibit proteolytic processing of apical membrane antigen 1 of Plasmodium falciparum merozoites. Proc Natl Acad Sci USA 2003;100:12295-12300.
- 14. Urquiza M, Suarez JE, Cardenas C, Lopez R, Puentes A, Chavez F, Calvo JC, Patarroyo ME.
Plasmodium falciparum AMA-1 erythrocyte binding peptides implicate AMA-1 as erythrocyte binding protein. Vaccine 2000;19:508-513.
- 15. Treeck M, Zacherl S, Herrmann S, Cabrera A, Kono M, Struck NS, Engelberg K, Haase S, Frischknecht F, Miura K, Spielmann T, Gilberger TW. Functional analysis of the leading malaria vaccine candidate AMA-1 reveals an essential role for the cytoplasmic domain in the invasion process. PLoS Pathog 2009;5:e1000322.
- 16. Sirima SB, Durier C, Kara L, Houard S, Gansane A, Loulergue P, Bahuaud M, Benhamouda N, Nebié I, Faber B, Remarque E, Launay O. Safety and immunogenicity of a recombinant Plasmodium falciparum AMA1-DiCo malaria vaccine adjuvanted with GLA-SE or Alhydrogel® in European and African adults: a phase 1a/1b, randomized, double-blind multi-centre trial. Vaccine 2017;35:6218-6227.
- 17. Payne RO, Milne KH, Elias SC, Edwards NJ, Douglas AD, Brown RE, Silk SE, Biswas S, Miura K, Roberts R, Rampling TW, Venkatraman N, Hodgson SH, Labbé GM, Halstead FD, Poulton ID, Nugent FL, de Graaf H, Sukhtankar P, Williams NC, Ockenhouse CF, Kathcart AK, Qabar AN, Waters NC, Soisson LA, Birkett AJ, Cooke GS, Faust SN, Woods C, Ivinson K, McCarthy JS, Diggs CL, Vekemans J, Long CA, Hill AV, Lawrie AM, Dutta S, Draper SJ. Demonstration of the blood-stage Plasmodium falciparum controlled human malaria infection model to assess efficacy of the P. falciparum apical membrane antigen 1 vaccine, FMP2.1/AS01. J Infect Dis 2016;213:1743-1751.
- 18. Sagara I, Dicko A, Ellis RD, Fay MP, Diawara SI, Assadou MH, Sissoko MS, Kone M, Diallo AI, Saye R, Guindo MA, Kante O, Niambele MB, Miura K, Mullen GE, Pierce M, Martin LB, Dolo A, Diallo DA, Doumbo OK, Miller LH, Saul A. A randomized controlled phase 2 trial of the blood stage AMA1-C1/Alhydrogel malaria vaccine in children in Mali. Vaccine 2009;27:3090-3098.
- 19. Thera MA, Coulibaly D, Kone AK, Guindo AB, Traore K, Sall AH, Diarra I, Daou M, Traore IM, Tolo Y, Sissoko M, Niangaly A, Arama C, Baby M, Kouriba B, Sissoko MS, Sagara I, Toure OB, Dolo A, Diallo DA, Remarque E, Chilengi R, Noor R, Sesay S, Thomas A, Kocken CH, Faber BW, Imoukhuede EB, Leroy O, Doumbo OK. Phase 1 randomized controlled trial to evaluate the safety and immunogenicity of recombinant Pichia pastoris-expressed Plasmodium falciparum apical membrane antigen 1 (PfAMA1-FVO [25-545]) in healthy Malian adults in Bandiagara. Malaria J 2016;15:442.
- 20. Laurens MB, Thera MA, Coulibaly D, Ouattara A, Kone AK, Guindo AB, Traore K, Traore I, Kouriba B, Diallo DA, Diarra I, Daou M, Dolo A, Tolo Y, Sissoko MS, Niangaly A, Sissoko M, Takala-Harrison S, Lyke KE, Wu Y, Blackwelder WC, Godeaux O, Vekemans J, Dubois MC, Ballou WR, Cohen J, Dube T, Soisson L, Diggs CL, House B, Bennett JW, Lanar DE, Dutta S, Heppner DG, Plowe CV, Doumbo OK. Extended safety, immunogenicity and efficacy of a blood-stage malaria vaccine in malian children: 24-month follow-up of a randomized, double-blinded phase 2 trial. PLoS One 2013;8:e79323.
- 21. Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum
. Nature 2002;419:498-511.
- 22. Thera MA, Doumbo OK, Coulibaly D, Laurens MB, Ouattara A, Kone AK, Guindo AB, Traore K, Traore I, Kouriba B, Diallo DA, Diarra I, Daou M, Dolo A, Tolo Y, Sissoko MS, Niangaly A, Sissoko M, Takala-Harrison S, Lyke KE, Wu Y, Blackwelder WC, Godeaux O, Vekemans J, Dubois MC, Ballou WR, Cohen J, Thompson D, Dube T, Soisson L, Diggs CL, House B, Lanar DE, Dutta S, Heppner DG Jr, Plowe CV. A Field Trial to Assess a Blood-Stage Malaria Vaccine. N Engl J Med 2011;365:1004-1013.
- 23. Al-Qahtani AA, Abdel-Muhsin AMA, Bin Dajem SM, AlSheikh AAH, Bohol MFF, Al-Ahdal MN, Putaporntip C, Jongwutiwes S. Comparative sequence analysis of domain I of Plasmodium falciparum apical membrane antigen 1 from Saudi Arabia and worldwide isolates. Infect Genet Evol 2016;39:381-388.
- 24. Basu M, Maji AK, Mitra M, Sengupta S. Natural selection and population genetic structure of domain-I of Plasmodium falciparum apical membrane antigen-1 in India. Infect Genet Evol 2013;18:247-256.
- 25. Garg S, Alam MT, Das MK, Dev V, Kumar A, Dash AP, Sharma YD. Sequence diversity and natural selection at domain I of the apical membrane antigen 1 among Indian Plasmodium falciparum populations. Malaria J 2007;6:154.
- 26. Mardani A, Keshavarz H, Heidari A, Hajjaran H, Raeisi A, Khorramizadeh MR. Genetic diversity and natural selection at the domain I of apical membrane antigen-1 (AMA-1) of Plasmodium falciparum in isolates from Iran. Exp Parasitol 2012;130:456-462.
- 27. Mehrizi AA, Sepehri M, Karimi F, Djadid ND, Zakeri S. Population genetics, sequence diversity and selection in the gene encoding the Plasmodium falciparum apical membrane antigen 1 in clinical isolates from the south-east of Iran. Infect Genet Evol 2013;17:51-61.
- 28. Rajesh V, Singamsetti VK, Vidya S, Gowrishankar M, Elamaran M, Tripathi J, Radhika NB, Kochar D, Ranjan A, Roy SK, Das A.
Plasmodium falciparum: genetic polymorphism in apical membrane antigen-1 gene from Indian isolates. Exp Parasitol 2008;119:144-151.
- 29. Arnott A, Wapling J, Mueller I, Ramsland PA, Siba PM, Reeder JC, Barry AE. Distinct patterns of diversity, population structure and evolution in the AMA-1 genes of sympatric Plasmodium falciparum and Plasmodium vivax populations of Papua New Guinea from an area of similarly high transmission. Malaria J 2014;13:233.
- 30. Cortes A, Mellombo M, Mueller I, Benet A, Reeder JC, Anders RF. Geographical structure of diversity and differences between symptomatic and asymptomatic infections for Plasmodium falciparum vaccine candidate AMA-1. Infect Immun 2003;71:1416-1426.
- 31. Tanabe K, Mita T, Palacpac NM, Arisue N, Tougan T, Kawai S, Jombart T, Kobayashi F, Horii T. Within-population genetic diversity of Plasmodium falciparum vaccine candidate antigens reveals geographic distance from a Central sub-Saharan African origin. Vaccine 2013;31:1334-1339.
- 32. Duan JH, Mu JB, Thera MA, Joy D, Pond SLK, Diemert D, Long C, Zhou H, Miura K, Ouattara A, Dolo A, Doumbo O, Su XZ, Miller L. Population structure of the genes encoding the polymorphic Plasmodium falciparum apical membrane antigen 1: implications for vaccine design. Proc Natl Acad Sci USA 2008;105:7857-7862.
- 33. Osier FH, Weedall GD, Verra F, Murungi L, Tetteh KK, Bull P, Faber BW, Remarque E, Thomas A, Marsh K, Conway DJ. Allelic diversity and naturally acquired allele-specific antibody responses to Plasmodium falciparum apical membrane antigen 1 in Kenya. Infect Immun 2010;78:4625-4633.
- 34. Ouattara A, Mu J, Takala-Harrison S, Saye R, Sagara I, Dicko A, Niangaly A, Duan J, Ellis RD, Miller LH, Su XZ, Plowe CV, Doumbo OK. Lack of allele-specific efficacy of a bivalent AMA-1 malaria vaccine. Malaria J 2010;9:175.
- 35. Ouattara A, Takala-Harrison S, Thera MA, Coulibaly D, Niangaly A, Saye R, Tolo Y, Dutta S, Heppner DG, Soisson L, Diggs CL, Vekemans J, Cohen J, Blackwelder WC, Dube T, Laurens MB, Doumbo OK, Plowe CV. Molecular basis of allele-specific efficacy of a blood-stage malaria vaccine: vaccine development implications. J Infect Dis 2013;207:511-519.
- 36. Polley SD, Conway DJ. Strong diversifying selection on domains of the Plasmodium falciparum apical membrane antigen 1 gene. Genetics 2001;158:1505-1512.
- 37. Soulama I, Bigoga JD, Ndiaye M, Bougouma EC, Quagraine J, Casimiro PN, Stedman TT, Sirima SB. Genetic diversity of polymorphic vaccine candidate antigens (apical membrane antigen-1, merozoite surface protein-3, and erythrocyte binding antigen-175) in Plasmodium falciparum isolates from western and central Africa. Am J Trop Med Hyg 2011;84:276-284.
- 38. Takala SL, Coulibaly D, Thera MA, Batchelor AH, Cummings MP, Escalante AA, Ouattara A, Traoré K, Niangaly A, Djimdé AA, Doumbo OK, Plowe CV. Extreme polymorphism in a vaccine antigen and risk of clinical malaria: implications for vaccine development. Sci Transl Med 2009;1:2ra5.
- 39. Tetteh KK, Stewart LB, Ochola LI, Amambua-Ngwa A, Thomas AW, Marsh K, Weedall GD, Conway DJ. Prospective Identification of Malaria Parasite Genes under Balancing Selection. PLoS One 2009;4:e5568.
- 40. Escalante AA, Grebert HM, Chaiyaroj SC, Magris M, Biswas S, Nahlen BL, Lal AA. Polymorphism in the gene encoding the apical membrane antigen-1 (AMA-1) of Plasmodium falciparum. X. Asembo Bay Cohort Project. Mol Biochem Parasitol 2001;113:279-287.
- 41. Ord RL, Tami A, Sutherland CJ. AMA1 genes of sympatric Plasmodium vivax and P. falciparum from Venezuela differ significantly in genetic diversity and recombination frequency. PLoS One 2008;3:e3366.
- 42. Tanabe K, Zollner G, Vaughan JA, Sattabongkot J, Khuntirat B, Honma H, Mita T, Tsuboi T, Coleman R.
Plasmodium falciparum: Genetic diversity and complexity of infections in an isolated village in western Thailand. Parasitol Int 2015;64:260-266.
- 43. Cui LW, Yan G, Sattabongkot J, Cao Y, Chen B, Chen X, Fan Q, Fang Q, Jongwutiwes S, Parker D, Sirichaisinthop J, Kyaw MP, Su XZ, Yang H, Yang Z, Wang B, Xu J, Zheng B, Zhong D, Zhou G. Malaria in the Greater Mekong Subregion: heterogeneity and complexity. Acta Trop 2012;121:227-239.
- 44. Imwong M, Suwannasin K, Kunasol C, Sutawong K, Mayxay M, Rekol H, Smithuis FM, Hlaing TM, Tun KM, van der Pluijm RW, Tripura R, Miotto O, Menard D, Dhorda M12, Day NPJ, White NJ, Dondorp AM. The spread of artemisinin-resistant Plasmodium falciparum in the Greater Mekong subregion: a molecular epidemiology observational study. Lancet Infect Dis 2017;17:491-497.
- 45. Polley SD, Chokejindachai W, Conway DJ. Allele frequency-based analyses robustly map sequence sites under balancing selection in a malaria vaccine candidate antigen. Genetics 2003;165:555-561.
- 46. Zhu X, Zhao Z, Feng Y, Li P, Liu F, Liu J, Yang Z, Yan G, Fan Q, Cao Y, Cui L. Genetic diversity of the Plasmodium falciparum apical membrane antigen I gene in parasite population from the China-Myanmar border area. Infect Genet Evol 2016;39:155-162.
- 47. Kang JM, Lee J, Moe M, Jun H, Lê HG, Kim TI, Thái TL, Sohn WM, Myint MK, Lin K, Shin HJ, Kim TS, Na BK. Population genetic structure and natural selection of Plasmodium falciparum apical membrane antigen-1 in Myanmar isolates. Malar J 2018;17:71.
- 48. Pumpaibool T, Arnathau C, Durand P, Kanchanakhan N, Siripoon N, Suegorn A, Sitthi-Amorn C, Renaud F, Harnyuttanakorn P. Genetic diversity and population structure of Plasmodium falciparum in Thailand, a low transmission country. Malaria J 2009;8:155.
- 49. Simpalipan P, Pattaradilokrat S, Siripoon N, Seugorn A, Kaewthamasorn M, Butcher RD, Harnyuttanakorn P. Diversity and population structure of Plasmodium falciparum in Thailand based on the spatial and temporal haplotype patterns of the C-terminal 19-kDa domain of merozoite surface protein-1. Malaria J 2014;13:54.
- 50. Sawaswong V, Simpalipan P, Siripoon N, Harnyuttanakorn P, Pattaradilokrat S. Allelic diversity and geographical distribution of the gene encoding Plasmodium falciparum merozoite surface protein-3 in Thailand. Korean J Parasitol 2015;53:177-187.
- 51. Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009;25:1451-1452.
- 52. Kumar S, Nei M, Dudley J, Tamura K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform 2008;9:299-306.
- 53. Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 1986;3:418-426.
- 54. Jukes TH, Cantor CR. Evolution of protein molecules. In Munro HN, editor. Mammalian Protein Metabolism. New York, USA. Academic Press; 1969. p. 21-132.
- 55. Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics 1993;133:693-709.
- 56. Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989;123:585-595.
- 57. Hudson RR, Kaplan NL. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 1985;111:147-164.
- 58. Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 2010;10:564-567.
- 59. Kocken CH, Narum1 DL, Massougbodji A, Ayivi B, Dubbeld MA, van der Wel A, Conway DJ, Sanni A, Thomas AW. Molecular characterisation of Plasmodium reichenowi apical membrane antigen-1 (AMA-1), comparison with P-falciparum AMA-1, and antibody-mediated inhibition of red cell invasion. Mol Biochem Parasitol 2000;109:147-156.
- 60. Conway DJ, Roper C, Oduola AM, Arnot DE, Kremsner PG, Grobusch MP, Curtis CF, Greenwood BM. High recombination rate in natural populations of Plasmodium falciparum
. Proc Natl Acad Sci USA 1999;96:4506-4511.
- 61. Kimura M. The Neutral Theory of Molecular Evolution. Cambridge, New York, USA. Cambridge University Press; 1983.
- 62. Coley AM, Campanale NV, Casey JL, Hodder AN, Crewther PE, Anders RF, Tilley LM, Foley M. Rapid and precise epitope mapping of monoclonal antibodies against Plasmodium falciparum AMA1 by combined phage display of fragments and random peptides. Protein Eng 2001;14:691-698.
- 63. Coley AM, Parisi K, Masciantonio R, Hoeck J, Casey JL, Murphy VJ, Harris KS, Batchelor AH, Anders RF, Foley M. The most polymorphic residue on Plasmodium falciparum apical membrane antigen 1 determines binding of an invasion-inhibitory antibody. Infect Immun 2006;74:2628-2636.
- 64. Kato K, Mayer DC, Singh S, Reid M, Miller LH. Domain III of Plasmodium falciparum apical membrane antigen 1 binds to the erythrocyte membrane protein Kx. Proc Natl Acad Sci USA 2005;102:5552-5557.
- 65. Ellis RD, Wu Y, Martin LB, Shaffer D, Miura K, Aebig J, Orcutt A, Rausch K, Zhu D, Mogensen A, Fay MP, Narum DL, Long C, Miller L, Durbin AP. Phase 1 study in malaria naïve adults of BSAM2/Alhydrogel®+CPG 7909, a blood stage vaccine against P. falciparum malaria. PLoS One 2012;7:e46094.
- 66. Laurens MB, Kouriba B, Bergmann-Leitner E, Angov E, Coulibaly D, Diarra I, Daou M, Niangaly A, Blackwelder WC, Wu Y, Cohen J, Ballou WR, Vekemans J, Lanar DE, Dutta S, Diggs C, Soisson L, Heppner DG, Doumbo OK, Plowe CV, Thera MA. Strain-specific Plasmodium falciparum growth inhibition among Malian children immunized with a blood-stage malaria vaccine. PLoS One 2017;12:e0173294.
- 67. Miura K, Herrera R, Diouf A, Zhou H, Mu J, Hu Z, MacDonald NJ, Reiter K, Nguyen V, Shimp RL Jr, Singh K, Narum DL, Long CA, Miller LH. Overcoming allelic specificity by immunization with five allelic forms of Plasmodium falciparum apical membrane antigen 1. Infect Immun 2013;81:1491-1501.