Abstract
Triatoma rubrofasciata is a wide-spread vector of Chagas disease in Americas. In this study, we completed the mitochondrial genome sequencing of T. rubrofasciata. The total length of T. rubrofasciata mitochondrial genome was 17,150 bp with the base composition of 40.4% A, 11.6% G, 29.4% T and 18.6% C. It included 13 protein-coding genes, 22 tRNA genes, 2 rRNA genes and one control region. We constructed a phylogenetic tree on the 13 protein-coding genes of T. rubrofasciata and other 13 closely related species to show their phylogenic relationship. The determination of T. rubrofasciata mitogenome would play an important role in understanding the genetic diversity and evolution of triatomine bugs.
-
Key words: Triatoma rubrofasciata, mitochondrial genome, protein-coding gene, phylogeny
Chagas disease, also known as American trypanosomiasis, caused by the protozoan parasite,
Trypanosoma cruzi, infects 8 million people worldwide, mostly in Latin America. In the past decades, it had been increasingly detected in the United States of America, Canada, and many European and some Western Pacific countries. This is mainly due to population mobility between Latin America and the rest of the world [
1].
This disease is transmitted mainly through the biting of triatomine (reduviid) bugs, also called as ‘kissing bug’. The subfamily Triatominae is divided into 149 species, that composed of 5 tribes: Alberproseniini, Bolboderini, Cavernicolini, Rhodniini and Triatomini, which is distributed from the southern USA to Patagonia, and a few species of Triatomini are in India and Australia [
2]. So far, information of only 2 mitochondrial (mt) genomes of triatomine bugs has been published. One is
Triatoma dimidiata, mainly distributed from Mexico to Northern South America [
3], the other is
Triatoma infestans, the main vector of Chagas disease in South America [
4]. In China, information of triatomine bugs was far from complete. Only a few reports show that
T. rubrofasciata and
Triatoma sinica are the 2 main native triatomine bugs [
5]. It was used to distribute in Fujian, Guangxi, Hainan and Taiwan province in l980s [
6,
7]. In 2017,
T. rubrofasciata from Guangdong province had been characterized by 16S rRNA sequence [
5]. However, complete information of its mt genome is still unknown.
For better understanding the biology of this species, the complete mt genome of T. rubrofasciata was determined and analyzed in this study. Furthermore, we constructed a phylogenetic tree on the 13 protein-coding genes of T. rubrofasciata and other 13 closely related species that show Triatoma species cluster together as a sister clade of Stenopodainae. Our findings will shed light on molecular taxonomy and population genetic studies of this important disease transmitting vector.
A single female adult of
T. rubrofasciata collected from the rural area of Baoting County (18°39′N 109°41′E) Hainan Province was used. The sample was stored in 100% ethanol at −20°C. The total DNA was extracted using the Holmes-Bonner buffer [
8] and stored at −20°C until use. The complete mt genome sequence of
T. rubrofasciata was amplified in 5 overlapping fragments, using 5 pairs of oligonucleotide primers designed based on the conserved regions from published complete mt genome of
T. dimidiata (NC_002609.1) (
Table 1). All PCR amplification reactions were carried out in a final volume of 25 μl, which included 9.5 μl of H
2O, 12.5 μl of 1–5
TM 2X High-Fidelity Master Mix (Molecular Cloning Laboratones MCLAB, South San Francisco, California, USA), 1 μl of each primer (10 μM), and 1 μl of DNA extract. Thermocycler conditions were as follows: 98°C for 2 min; 25 to 35 cycles of 98°C for 10 sec, 45–68°C for 15 sec, 72°C for 30–90 sec depending on fragment lengths, followed by 5–15 s/kb, then a final extension at 72°C for 5 min. These amplicons were sequenced by Shanghai Tsingke (Shanghai, China) using primer walking in both directions.
The annotation of mt genome for
T. rubrofasciata was preformed using web-based services MITOS (
http://mitos.bioinf.uni-leipzig.de/help.py) [
9]. Protein-coding genes were annotated by checking manually for consistent start/stop codons, open reading frames. Then, tRNAs were identified by the proposed clover-leaf secondary structure and predicted using web-based tRNA-scan SE 1.21 program (
http://lowelab.ucsc.edu/tRNAscan-SE/) [
10] with default search mode. The rRNA genes were annotated by extending until adjacent tRNAs [
11]. Comparing with other mt genomes, the 5′ end of the srRNA gene was confined. Moreover, the base composition and codon usage were assessed using MEGA version 6.0 [
12]. Finally, the graphical map of mt genome was drawn using web-based OrganellarGenomeDRAW (
http://ogdraw.mpimp-golm.mpg.de/) [
13] (
Fig. 1).
Previously published mt genome under other Reduvidae species were obtained from GenBank, including
Brontostoma colossus (KM044501),
Oncocephalus breviscutum (KC887527),
Peirates arcuatus (KF752445),
Peirates atromaculatus (KF913538),
Peirates fulvescens (KF913537),
Peirates lepturoides (KF913541),
Peirates turpis (KF913540),
Sirthenea flavipes (HQ645959),
T. dimidiata (AF301594),
T. infestans (KY640305), and
Valentia hoffmanni (FJ456952). Furthermore, the mt genome from
Apolygus lucorum (HQ902161) and
Corythucha ciliate (KC756280) were used as outgroup. Then, protein-coding genes were aligned using ClustalW in MEGA 6.0 [
12] with default settings. Thus, the sequences from the 13 protein-coding genes alignments were concatenated to single multiple sequence alignment for phylogenetic analysis. The phylogenetic relationships were constructed using maximum-likelihood method in MEGA 6.0, with node support was assessed with 1,000 bootstrap replicates (
Fig. 2).
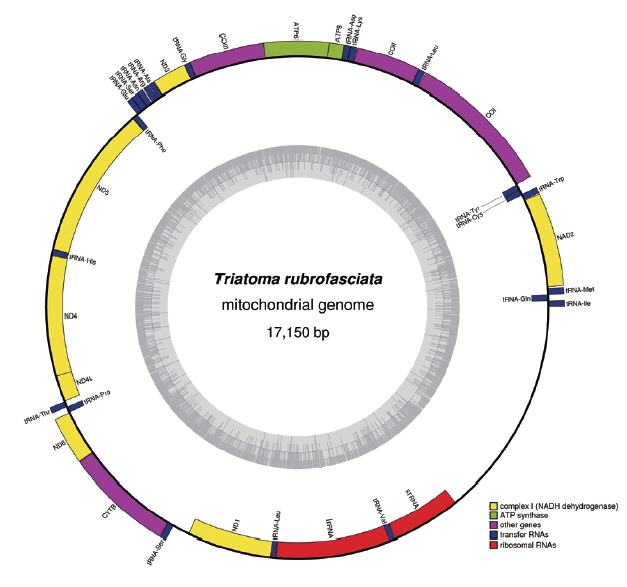
The complete mt genome of
T. rubrofasciata was a typically circular molecule with 17,150 bp in length (GenBank accession no. MH934953). The size of
T. rubrofasciata mt genome was smaller than
T. infestans (17,301 bp) [
4], whereas was larger than
T. dimidiata (17,019 bp) [
3]. The slight difference in length is mainly due to the repeat region. The mt genome of
T. rubrofasciata consisted of 13 protein-coding genes, 22 transfer RNAs, 2 ribosomal RNAs and a control region (
Table 2). The genome organization was consistent with
T. infestans, T. dimidiata and other reduviid genomes, such as
Sirthenea flavipes [
14] and
Oncocephalus breviscutum [
15]. However, there was 23 tRNA instead of 22 in
Brontostoma Colossus [
16]. Moreover, heavy strand (H strand) encoded 23 genes, whereas light strand (L strand) encoded the remaining 14 genes, and the overlap sequences were found at 16 locations. Most overlap sequence was only 1 bp and the longest overlap was 15 bp between ND1 and tRNA-Leu. In
T. rubrofasciata, several intergenic sequences were observed, however the intergenic spacer function was still unknown. The most longest intergenic spacer located between tRNA-Ser and ND1 was similar to the intergenic spacer of
Apis mellifera that thought to be another origin of replication [
3]. There was a bias toward A and T nucleotides in mt genome nucleotide composition. The overall nucleotide composition of H-strand was 34.9% A, 34% T, 12.9% G, and 18.2% C, thus biased toward A+T (68.9%).
The canonical start codons of protein-coding genes were ATN, GTG and TTG in invertebrate [
17]. In
T. rubrofasciata, the start codon of 13 protein-coding genes was ATN, instead of GTG and TTG. The start codons for most of the coding sequences was ATT (ND2, COII, ND5, ND1) and ATG (COI, ATP6, COIII, ND4, ND4L, ND6, CytB), except that ATC for ATP8 and ATA for ND3. Almost all start codons of protein-coding genes were consist with
T. infestans and
T. dimidiata. However, in
T. rubrofasciata, ND5 start codons was ATT instead of GTG that was found in
T. infestans and
T. dimidiata. Most coding sequences was terminated with TAA (ATP8, ATP6, ND3, ND5, ND4, ND4L, ND6, ND1). The stop codons of ND2 and CytB were TAG. The COI and COII were stopped with incomplete T (aa) stop codons, and COIII stop codon was TA (a). The incomplete TA (a) and T (aa) stop codons were the deletion version of canonical TAA stop codon. It was common in metazoan mt genomes [
11].
The 22 tRNA genes typically found in metazoan mt genomes were also identified in
T. rubrofasciata ranging in length from 61 bp to 73 bp (
Table 2). Most tRNA genes had the typical secondary structures except tRNA-Ser (AGN). It may be due to the formation of the dihydrouridine arm. The large rRNA subunit gene (lrRNA) was 1,266 bp long and the A+T content was 75.2%. The small rRNA subunit gene (srRNA) was 771 bp long and the A+T content was 70.8%.
The control region in
T. rubrofasciata was 2,428 bp in length. It was smaller than
T. infestans (2,653 bp), whereas was larger than
T. dimidiata (2,166 bp). The control region was the most variable region between Reduviidae species [
16]. This region in
T. rubrofasciata was divided into 4 different parts. The first part was 467 bp in length and located downstream of srRNA gene and ended with 11 guanines. The second region (436 bp) was located next to first region, heavily biased toward A+T (78.4%). The third region was the longest region (1,357 bp) and composed of 2 different tandem repeat units. One was 760 bp in length with 5 consecutive 152 bp repetitions. The other had 2 consecutive 198 bp repetitions. The fourth part of the control region was 168 bp, ended at the beginning of the tRNA-Ile, which had the potential to form stem-and-loop structure.
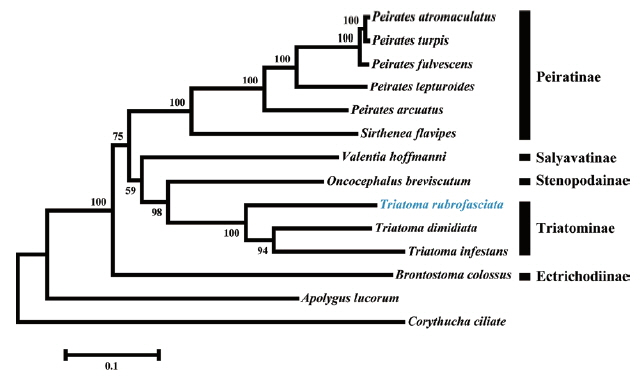
We constructed a phylogenetic tree based on the analysis of 13 protein-coding genes (
Fig. 2). As expected,
T. rubrofasciata was clustered together with
T. dimidiata and
T. infestans that grouped in a highly supported clade.
Triatoma species and
Oncocephalus breviscutum (Stenopodainae) also grouped together in an extremely close clade. The close relationship between both subfamilies had been confirmed using morphological characteristics and phylogenetic analyses of the mitochondria 16S and nuclear 18S and 28S ribosomal DNA gene sequences [
18,
19].
In conclusion, our study details the complete sequencing and annotating of the mt genome of T. rubrofasciata. The complete mt genome contains 37 genes, including 13 protein-coding genes, 2 ribosomal RNA genes and 22 transfer RNA genes. Phylogenetic analyses using the amino acid sequences of the 13 protein-coding genes showed that the Triatoma species cluster together as a sister clade of Stenopodainae. This mt genome provides a unique genetic marker for studying the molecular biology, genetics and systematics of T. rubrofasciata.
Notes
-
CONFLICT OF INTEREST
There is no conflict of interest related to this work.
ACKNOWLEDGMENT
This study was supported by the “National Research and Development Plan of China” (No. 2016YFC1200500).
Fig. 1Graphical map of the complete mitochondrial genome of Triatoma rubrofasciata. Genes encoded by the heavy strand were shown outside the circle, and genes encoded by the light strand were shown inside the circle, respectively. The GC content of the genome were shown in the inner circle.
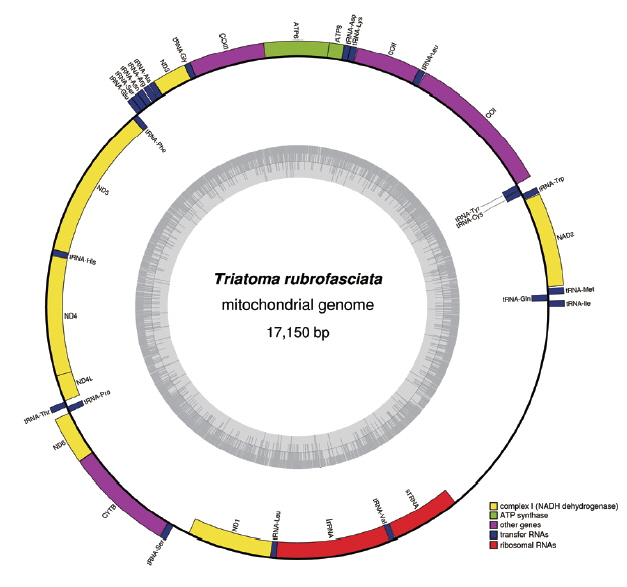
Fig. 2Phylogenetic tree based on Maximum likelihood analysis of 13 protein-coding genes. Sequence from the present study was indicated with a blue font. Apolygus lucorum and Corythucha ciliate were used as outgroup. Bootstrap support values were displayed at each node.
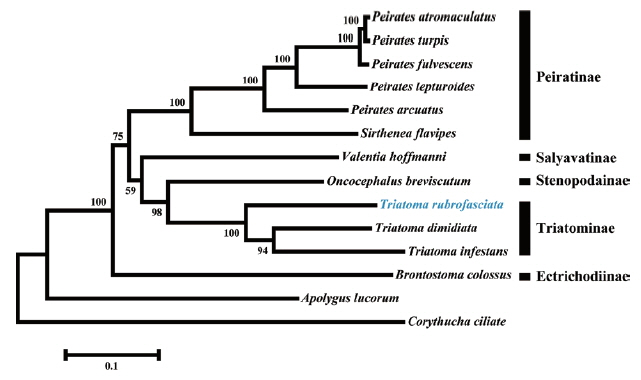
Table 1Primer sequences used to amplify PCR fragments of Triatoma rubrofasciata
Table 1
|
Primer name |
Sequences (5′-3′) |
Size (kb) |
|
Tr-1 |
ACCGCCTATTAATTCAGCCACTT
GCGTGTTCTTAGTCGAAGACTTGTT |
~7k |
|
Tr-2 |
ACACCCGCAGTAACCAAAGTAGAAG
AGGATGTAAGGTTCTTCAGCAGGAC |
~4k |
|
Tr-3 |
ACCATCTCGTCCGTTATCTTCTTCT
GCGGTTATACAATAGGAGCGAGTGA |
~4k |
|
Tr-4 |
AAACGAGAGTGACGGGCGATATG
GTTCATCCTGTTCCTGCTCCTCTT |
~4k |
|
Tr-5 |
TGGAAATGATGTCTTGGTTGCTA
TCAGAAAGACCTATGTACCTAAGAA |
~2k |
Table 2Annotation of the complete mitochondrial genome of
Triatoma rubrofasciata. tRNA abbreviations follow the IUPAC-IUB three letter code. For other abbreviation see legend for
Fig. 1.
Table 2
|
Gene |
Stand |
Nucleotide number |
Anticodon |
Start codon |
Stop codon |
|
tRNA-Ile |
H |
1-67 |
33-35 GAT |
- |
- |
|
tRNA-Gln |
L |
65-133 |
101-103 TTG |
- |
- |
|
tRNA-Met |
H |
133-200 |
163-165 CAT |
- |
- |
|
ND2 |
H |
219-1199 |
- |
ATT |
TAG |
|
tRNA-Trp |
H |
1206-1271 |
1236-1238 TCA |
- |
- |
|
tRNA-Cys |
L |
1264-1325 |
1293-1295 GCA |
- |
- |
|
tRNA-Tyr |
L |
1327-1392 |
1358-1360 GTA |
- |
- |
|
COI |
H |
1394-2922 |
- |
ATG |
T(aa) |
|
tRNA-Leu |
H |
2928-2996 |
2961-2963 TAA |
- |
- |
|
COII |
H |
2997-3672 |
- |
ATT |
T(aa) |
|
tRNA-Lys |
H |
3676-3745 |
3706-3708 CTT |
- |
- |
|
tRNA-Asp |
H |
3745-3808 |
3775-3777 GTC |
- |
- |
|
ATP8 |
H |
3809-3967 |
- |
ATC |
TAA |
|
ATP6 |
H |
3961-4644 |
- |
ATG |
TAA |
|
COIII |
H |
4631-5416 |
- |
ATG |
TA(a) |
|
tRNA-Gly |
H |
5416-5478 |
5446-5448 TCC |
- |
- |
|
ND3 |
H |
5476-5832 |
- |
ATA |
TAA |
|
tRNA-Ala |
H |
5833-5894 |
5862-5864 TGC |
- |
- |
|
tRNA-Arg |
H |
5898-5961 |
5927-5929 TCG |
- |
- |
|
tRNA-Asn |
H |
5973-6036 |
6003-6005 GTT |
- |
- |
|
tRNA-Ser(AGN) |
H |
6036-6108 |
6060-6062 GCT |
- |
- |
|
tRNA-Glu |
H |
6108-6168 |
6137-6139 TTC |
- |
- |
|
tRNA-Phe |
L |
6171-6234 |
6201-6203 GAA |
- |
- |
|
ND5 |
L |
6234-7946 |
- |
ATT |
TAA |
|
tRNA-His |
L |
7944-8009 |
7975-7977 GTG |
- |
- |
|
ND4 |
L |
8012-9340 |
- |
ATG |
TAA |
|
ND4L |
L |
9334-9627 |
- |
ATG |
TAA |
|
tRNA-Thr |
H |
9630-9692 |
9660-9662 TGT |
- |
- |
|
tRNA-Pro |
L |
9693-9757 |
9626-9628 TGG |
- |
- |
|
ND6 |
H |
9761-10261 |
- |
ATG |
TAA |
|
CytB |
H |
10261-11391 |
- |
ATG |
TAG |
|
tRNA-Ser(UCN) |
H |
11393-11459 |
11422-11424 TGA |
- |
- |
|
ND1 |
L |
11641-12573 |
- |
ATT |
TAA |
|
tRNA-Leu |
L |
12559-12624 |
12593-12595 TAG |
- |
- |
|
lrRNA |
L |
12625-13890 |
- |
- |
- |
|
tRNA-Val |
L |
13879-13949 |
13915-13917 TAC |
- |
- |
|
srRNA |
L |
13952-14722 |
- |
- |
- |
|
Control region |
|
14723-17150 |
- |
- |
- |
References
- 1. WHO. Chagas disease (American Trypanosomiasis). [internet]; 2018. Available from: http://www.who.int/mediacentre/fact-sheets/fs340/en/index.html
- 2. Schofield CJ, Galvão C. Classification, evolution, and species groups within the Triatominae. Acta Trop 2009;110:88-100.
- 3. Dotson EM, Beard CB. Sequence and organization of the mitochondrial genome of the Chagas disease vector, Triatoma dimidiata
. Insect Mol Biol 2001;10:205-215.
- 4. Pita S, Panzera F, Vela J, Mora P, Palomeque T, Lorite P. Complete mitochondrial genome of Triatoma infestans (Hemiptera, Reduviidae, Triatominae), main vector of Chagas disease. Infect Genet Evol 2017;54:158-163.
- 5. Liu Q, Guo YH, Zhang Y, Zhou ZB, Zhang LL, Zhu D, Zhou XN. First records of Triatoma rubrofasciata (De Geer, 1773) (Hemiptera, Reduviidae) in Foshan, Guangdong Province, Southern China. Infect Dis Poverty 2017;6:129.
- 6. Chen MX. The prevention of triatomine causing dermatitis. Hainan Med J 1986;35.
- 7. Cross JH, Hsu MY, Hung CK. Studies on trypanosomes in the Taiwan monkey. Southeast Asian J Trop Med Public Health 1983;14:536-542.
- 8. Holmes DS, Bonner J. Preparation, molecular weight, base composition, and secondary structure of giant nuclear ribonucleic acid. Biochemistry 1973;12:2330-2238.
- 9. Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol 2013;69:313-319.
- 10. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997;25:955-964.
- 11. Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981;290:470-474.
- 12. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 2013;30:2725-2729.
- 13. Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet 2007;52:267-274.
- 14. Gao JY, Li H, Truong XL, Dai X, Chang J, Cai WZ. Complete nucleotide sequence and organization of the mitochondrial genome of Sirthenea flavipes (Hemiptera: Reduviidae: Peiratinae) and comparison with other assassin bugs. Zootaxa 2013;3669:1-16.
- 15. Li H, Gao J, Cai W. Complete mitochondrial genome of the assassin bug Oncocephalus breviscutum (Hemiptera: Reduviidae). Mitochondrial DNA 2015;26:674-675.
- 16. Kocher A, Kamilari M, Lhuillier E, Coissac E, Péneau J, Chave J, Murienne J. Shotgun assembly of the assassin bug Brontostoma colossus mitochondrial genome (Heteroptera, Reduviidae). Gene 2014;552:184-194.
- 17. Cameron SL, Lo N, Bourguignon T, Svenson GJ, Evans TA. A mitochondrial genome phylogeny of termites (Blattodea: Termitoidae): Robust support for interfamilial relationships and molecular synapomorphies define major clades. Mol Phylogenet Evol 2012;65:163-173.
- 18. Weirauch C. Cladistic analysis of Reduviidae (Heteroptera: Cimicomorpha) based on morphological characters. Syst Entomol 2008;33:229-274.
- 19. Weirauch C, Munro JB. Molecular phylogeny of the assassin bugs (Hemiptera: Reduviidae), based on mitochondrial and nuclear ribosomal genes. Mol Phylogenet Evol 2009;53:287-299.