Abstract
Toxoplasma gondii is an obligate intracellular protozoan parasite that can invade various organs in the host body, including the central nervous system. Chronic intracranial T. gondii is known to be associated with neuroprotection against neurodegenerative diseases through interaction with host brain cells in various ways. The present study investigated the neuroprotective effects of chronic T. gondii infection in mice with cerebral ischemia experimentally produced by middle cerebral artery occlusion (MCAO) surgery. The neurobehavioral effects of cerebral ischemia were assessed by measurement of Garcia score and Rotarod behavior tests. The volume of brain ischemia was measured by triphenyltetrazolium chloride staining. The expression levels of related genes and proteins were determined. After cerebral ischemia, corrected infarction volume was significantly reduced in T. gondii infected mice, and their neurobehavioral function was significantly better than that of the uninfection control group. Chronic T. gondii infection induced the expression of hypoxia-inducible factor 1-alpha (HIF-1α) in the brain before MCAO. T. gondii infection also increased the expression of vascular endothelial growth factor after the cerebral ischemia. It is suggested that chronic intracerebral infection of T. gondii may be a potential preconditioning strategy to reduce neural deficits associated with cerebral ischemia and induce brain ischemic tolerance through the regulation of HIF-1α expression.
-
Key words: Toxoplasma gondii, cerebral ischemia, ischemic tolerance, neuroprotection
INTRODUCTION
Toxoplasma gondii is a globally occurring, intracellular protozoan parasite that can invade various organs of warm-blooded animals, including humans [
1]. Humans are infected through oocysts, tachyzoites or bradyzoites (tissue cysts) in contaminated food or water [
2]. Organ transplantation from an infected donor and vertical transmission from mother to fetus [
3] are other routes of
T. gondii infection in humans.
T. gondii tachyzoites actively penetrate nucleated host cells and enter the circulation, with dissemination to secondary tissues [
4]. After the acute phase of infection, the tachyzoites convert to bradyzoites and establish a chronic infection in various organs and tissues [
5]. It is well known that the brain is the most common site for latent
T. gondii infection [
4]. The intracellular parasites cross the blood-brain barrier by a Trojan horse mechanism and migrate to the brain as early as 7 days after infection [
6]. Initially, the parasites activate resident microglia and astrocytes in the brain and these recruit immune cells and produce proinflammatory cytokines to eliminate
T. gondii [
7]. However, the brain has immune privilege for
T. gondii infection since it displays different strategy in the course of infection in neural cells [
6]. Neurons do not have specific intracellular mechanisms for inhibiting
T. gondii growth and infected neurons are not recognized by CD8+ T cells because of the lack of MHC class I [
6]. The parasite lives in the host's brain in the form of bradyzoite-filled cysts after stage conversion [
8].
Neurons are fundamental elements of the central nerve system (CNS). Thus,
T. gondii cysts in neurons can directly or indirectly influence neuronal function and structure, potentially leading to altered host CNS function. Many studies have assessed the impact of chronic
T. gondii infection on host behavior in various rodent models [
9]. The hallmark of chronic stage of infection is the permanent resident glial cell activation in the brain. This ongoing low-level neuroinflammation is associated with these behavioral changes [
10]. Neuroinflammation is one of the main mediators of the pathophysiology of various CNS diseases and it may have both protective and deleterious effects in a disease [
11,
12]. Therefore, chronic
T. gondii infection in the brain might modify the course of various brain disorders. In our previous study, it was shown that chronic
T. gondii infection inhibited neuronal degeneration in a rodent model of Alzheimer’s disease [
13].
Immune responses also play an important role in lesion development, progression, and repair, and in recovery and overall outcome following ischemic stroke [
12]. In another study, a 40% to 50% decrease in infarct size was evident in chronic
T. gondii infections in a mouse model of permanent middle cerebral artery occlusion (MCAO) [
14]. Authors suggested that increased basal levels of anti-inflammatory cytokines and upregulated glutathione (GSH) were involved in the neuroprotective effect [
14]. However, the definitive mechanism of the neuroprotective effect in cerebral ischemia has not been established, and behavioral improvement after the ischemic stroke in
T. gondii infected mice has not been explored. We hypothesized that
T. gondii infection can increase ischemic tolerance in the brain, which leads to reduced infarct size and improvement of the functional deficit due to the ischemic stroke. In the study, we explored the neuroprotective effects of chronic
T. gondii infection in a rodent cerebral ischemia model and investigated the mechanism of protection of the brain tissue from ischemic damages.
A total of 36 ICR mice (7 weeks old; weight, 30–33 g; ORIENT BIO Inc., Gyeonggi-do, Korea) were used and divided into infection group (n=18) and control group (n=18). Tissue cysts isolated from the brains of
T. gondii ME49 infected mice were intraperitoneally injected into each of the mice in the infection group. Eighteen mice treated with PBS comprised the control group. The experimental mice were raised under specific pathogen-free conditions at the Biomedical Research Institute, Seoul National University Hospital Seoul, Korea. Body temperature and body weight were measured every 3 days to observe the systemic response due to the infection. Cerebral ischemia was surgically induced 4 weeks after the infection. The 90 min transient MCAO model of cerebral ischemia was a modification of a prior method [
15]. Three days after the surgery, mice were sacrificed, and the brains were harvested. Infections were confirmed retrospectively by microscopic observations of
T. gondii cysts in homogenized brain tissue after euthanasia.
The experimental protocol was approved by the Institutional Animal Care and Use Committee of Seoul National University Hospital (approval no. 15-0192).
The neurobehavioral effects of cerebral ischemia were assessed by measurement of the Garcia score and Rotarod test on days 1 and 3 after the surgery. The Garcia scale is a neurological score for cerebral ischemia in rodents [
16]. The maximum score of 18 points is determined by examining spontaneous activity, symmetry in the movement of limbs, forepaw outstretching, climbing, body proprioception, and response to vibrissae touch. The Rotarod test evaluates motor coordination and balance function of MCAO rodent models [
17]. In this test, each mouse was placed on a rotating cylinder and the speed of the cylinder was gradually increased (2.5 rotations per min [rpm] every 30 sec) from 2.5 to 40 rpm after a 5 min adaptation period. The maximum rpm was recorded, and the highest rpm from 3 repetitions indicated the motor coordination and balance function of the mice. On day 3, after behavioral tests, each mouse was euthanized in a chamber pre-filled with carbon dioxide, and their brains were harvested. The brains were sectioned coronally into 6 slices (1 mm thick) from the frontal pole. The second and fourth sections from the frontal pole were used for molecular analysis, and the remaining sections were used to measure the infarct volume (
Supplementary Fig. 1). All remaining brain tissue was used to confirm
T. gondii infection. Cerebral infarctions were identified using triphenyl tetrazolium chloride (TTC) staining and scanned into digital images [
18]. The infarct volume was calculated by a blinded tester using ImageJ software (NIH, Bethesda, Maryland, USA) based on the trapezoidal rule. Corrected infarct volume excluding brain edema [
19] was estimated. The ratios of the infarct volume to the volume of the contralateral hemisphere and ipsilateral hemisphere were also calculated.
Isolated brain tissues were also analyzed to determine the change in the gene expression of hypoxia-inducible factor 1-alpha (HIF-α and vascular endothelial growth factor (VEGF) to assess the protective effects of T. gondii chronic infection. Total RNA was extracted from the brain tissues using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The RNA concentration was determined using a NanoDrop One spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA). The cDNA was synthesized from RNA with a PrimeScript™ RT Master Mix (TaKaRa Bio Inc., Shiga, Japan). Quantitative real-time PCR was performed using the iQ5 real-time PCR detection system (Bio-Rad, Hercules, California, USA) using primers and templates in the SsoAdvanced™ Universal SYBR Green Supermix (Bio-Rad). DNA was amplified for 40 cycles of denaturation for 15 sec at 95°C and annealing for 30 sec at 60°C. The data generated from each PCR reaction were analyzed using the Data Analysis Module by iQTM5 optical system software (Bio-Rad). The specificity of reactions was determined by melting curve analysis. The relative fold-changes of gene expression between selected mRNAs and β-actin were calculated by the standard curve method.
Brain tissues were homogenized in a solution containing a protease inhibitor cocktail (Roche, Indianapolis, Indiana, USA) using a Potter-Elvehjem homogenizer (OMNI International, Kennesaw, Georgia, USA). Homogenates were centrifuged at 1,000 g for 15 min. Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. The membranes were then blocked with 5% skim milk in Tris-buffered saline for 2 hr. Protein bands were confirmed with HIF-α antibody (#ab1; AbCam, Cambridge, Massachusetts, USA) and horseradish peroxidase-conjugated secondary antibody (Amersham Pharmacia, Buckinghamshire, UK). Immune-reactive bands were visualized using an enhanced chemiluminescence system (ECL; Amersham Pharmacia, UK). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the loading control. Statistical analysis was conducted using an unpaired t-test. P-values<0.05 were considered significant. The results are presented as the mean±SD.
On days 1 and 3 after MCAO, hypothermia was observed to lower body temperature by approximately 1.5°C. However, there was no significant difference in body temperature and weight between the 2 groups during the entire experiment (data not shown). Postmortem brain analysis to confirm T. gondii infections in the infection group revealed cysts in all infected mice. Among the 18 mice in each group, 8 survived in the control group and 11 survived in the infection group 3 days after MCAO surgery. Two mice of each group were excluded from the analysis because autopsy showed that cerebral ischemia by the surgery was not adequately developed. Thus, 6 mice in the control group and 9 mice in the infection group were included in the data analysis.
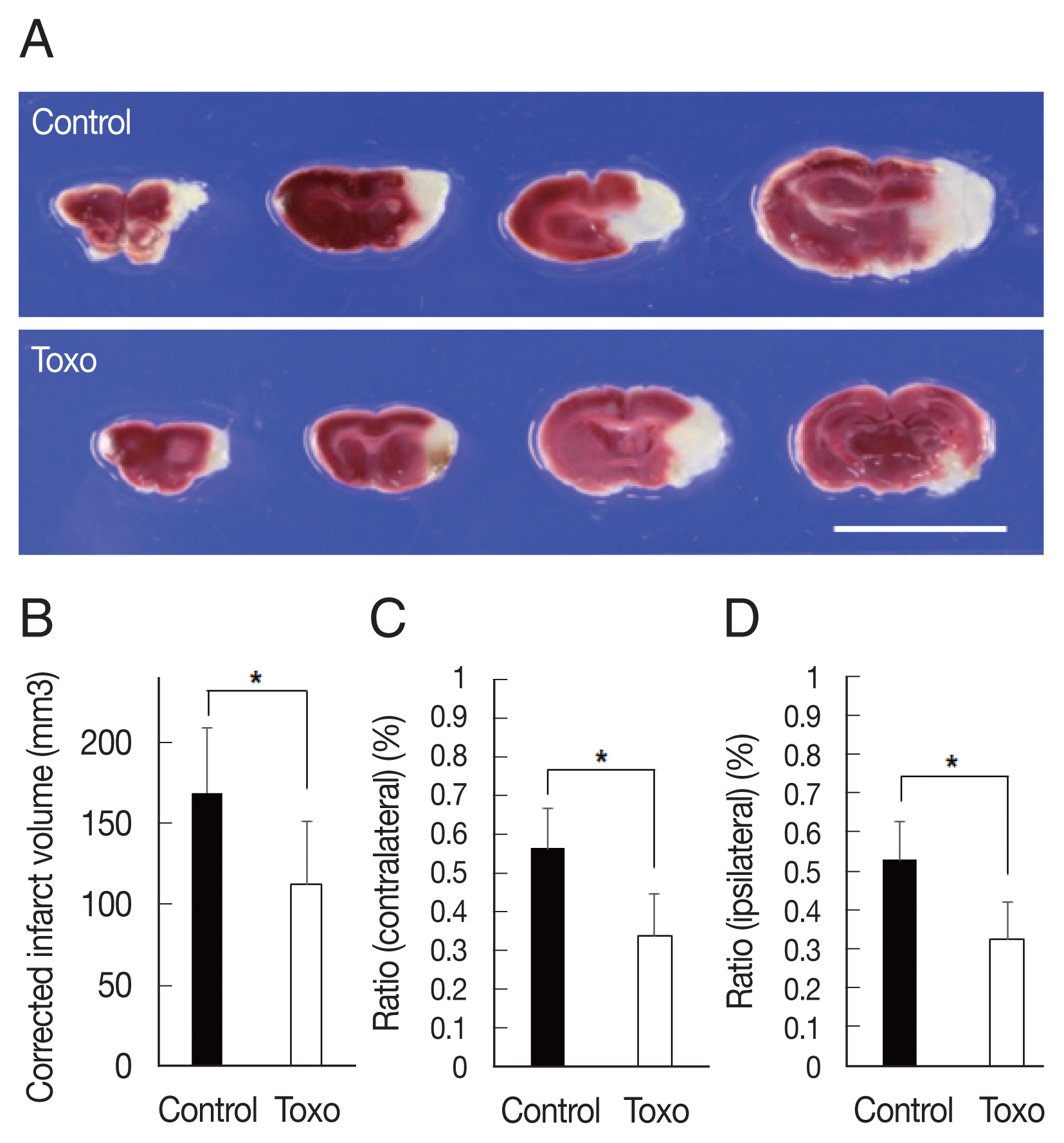
Corrected infarct volume measured from TTC staining was significantly decreased in the infection group (control, n=6, 168.8±40.4; infected, n=9, 112.5±39.2,
P=0.018). In the infection group, significant decreases were also evident in the ratios of the infarct volume to the contralateral hemisphere (control, n=6, 0.56±0.10; infected, n=9, 0.34±0.11,
P=0.001) and ipsilateral hemisphere (control, n=6, 0.53±0.10; infected, n=9, 0.33±0.10,
P=0.002) (
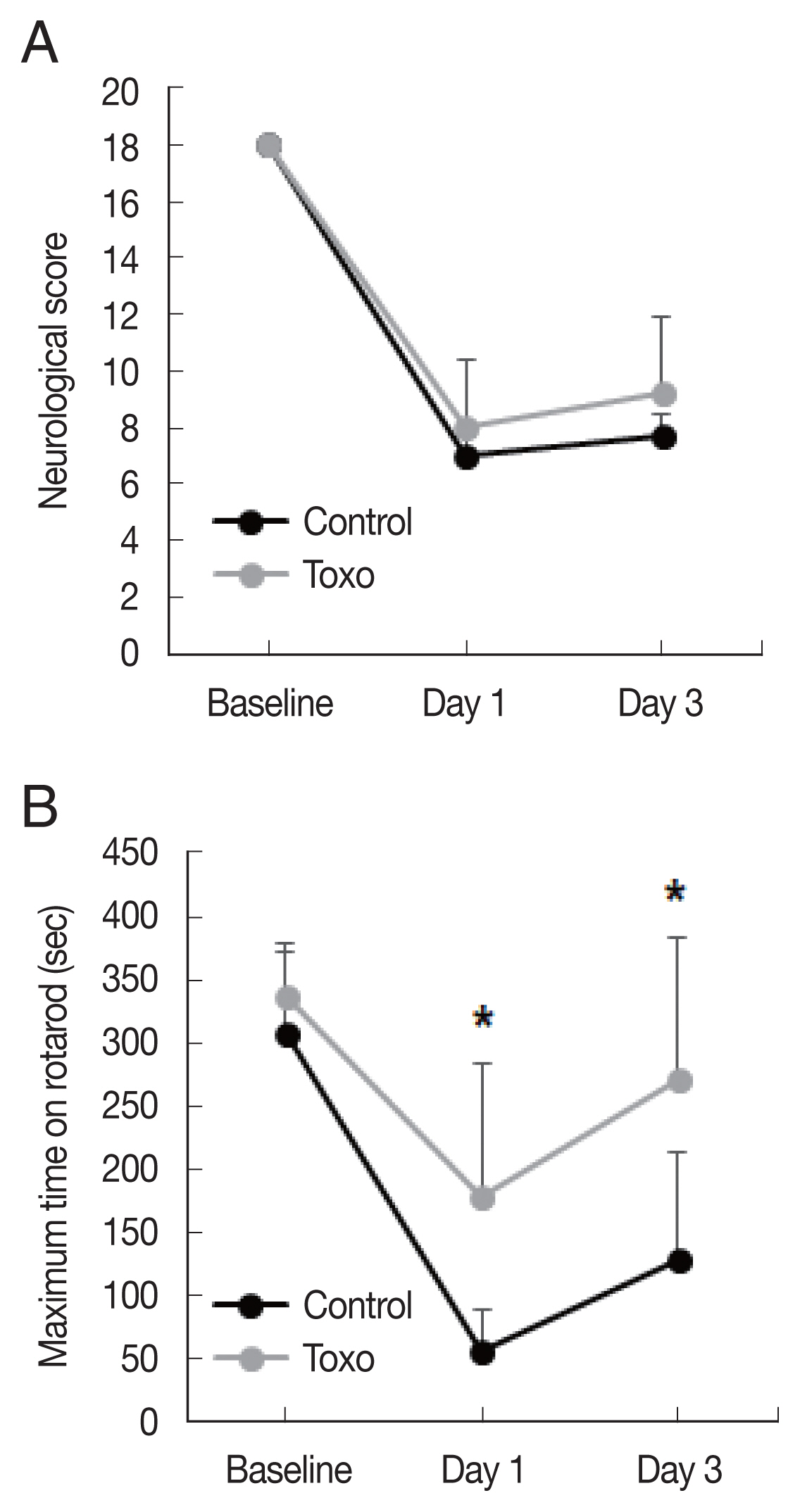
Fig. 1). The Garcia score in the infection group was slightly higher on both day 1 (control, n=6, 7.0±0.6; infected, n=9, 8.0±2.3,
P=0.253) and day 3 (control, n=7.7±0.8; infected, n=9, 9.2±2.7,
P=0.134) than that in the control group. The differences were not statistically significant. A significantly higher maximum time in the rotarod test in the infection group was observed on both day 1 (control, n=6, 55.0±33.0; infected, n=9, 178.1±105.8,
P=0.08) and day 3 (control, n=6, 127.2±85.4; infected, n=9, 271.6±112.1,
P= 0.019) (
Fig. 2).
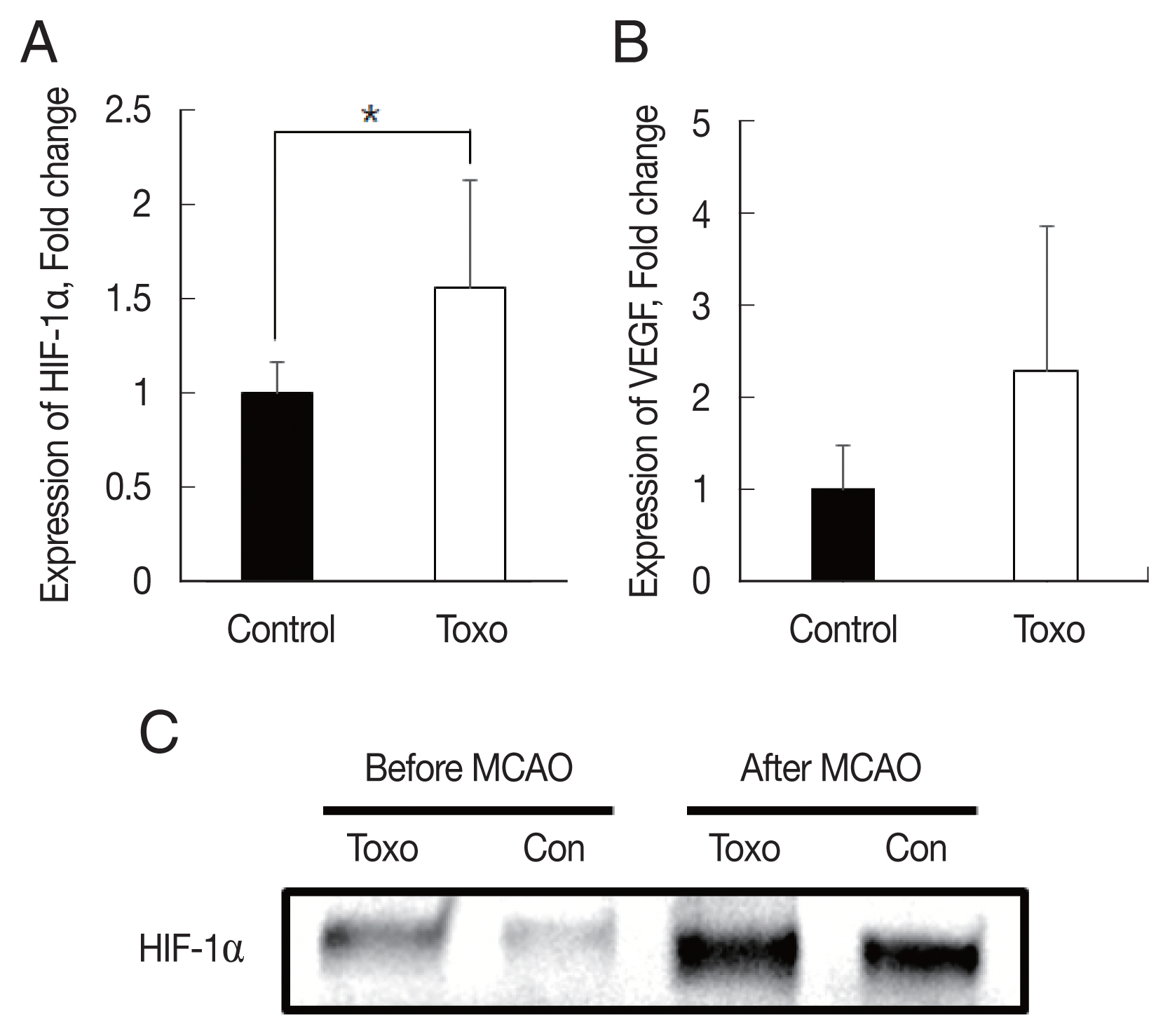
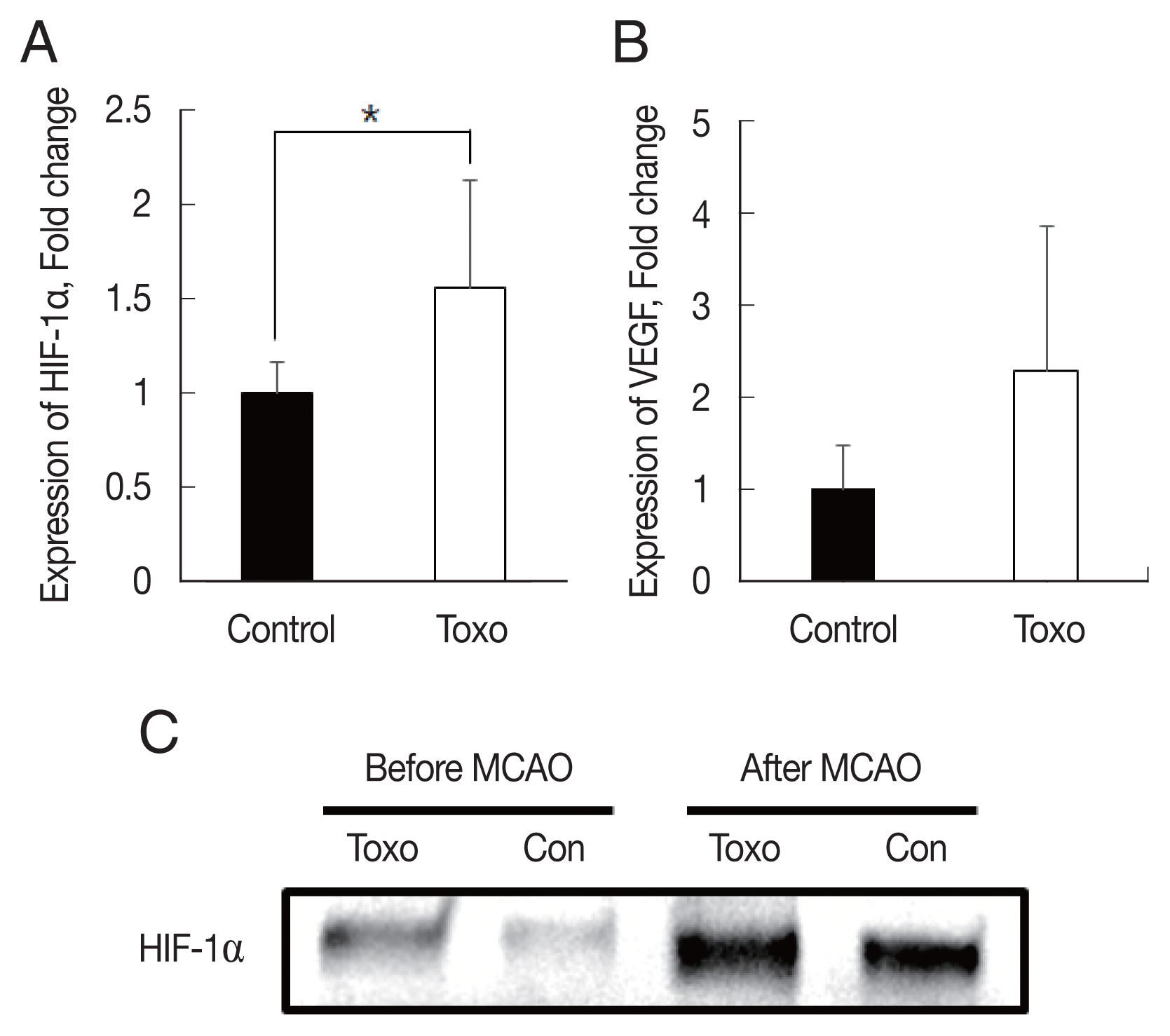
We determined whether
T. gondii infection induced brain ischemic tolerance through the regulation of preconditioning factors, such as HIF-1α and VEGF, in the brain of infected mice using qRT-PCR and western blotting (
Fig. 3). Expression levels of HIF-1α (
P<0.05) and VEGF were increased in the
T. gondii infection group (n=9) compared to those in the control group (n=6) after MCAO. These results indicated that infection of
T. gondii increased HIF-1α and VEGF expression in the host brain (
Fig. 3).
Our results revealed that chronic
T. gondii infection was neuroprotective in the ischemic stroke mouse model through behavior tests and induction of HIF-1α gene and VEGF protein. Infected mice showed ameliorated neurologic deterioration, better motor coordination, and decreased infarct size after the experimental cerebral ischemia. These findings are similar with those of a previous study that investigated the effect of chronic
T. gondii infection in cerebral ischemia [
14]. The previous study showed that infarct volume in infected mice was 40–50% that of the control, while it was 67% in the present study. We used a 90 min reperfusion MCAO and detected cerebral infarction with TTC staining. The previous study used permanent MCAO and stained brain slices with cresyl violet. Even though the infarct volume reduction in our study was smaller than that in the previous study, the results of the behavioral test reinforced the neuroprotective effect of chronic
T. gondii infection. The Garcia test revealed better neurological function in the infection group than the control group, although there was no statistical significance (day 1,
P=0.253; day 2,
P=0.134). Motor coordination and balance function recovery after cerebral ischemia in the rotarod test was significantly improved in the infection group. This could reflect the neuroprotection from the chronic
T. gondii infection. Reduction of the infarct size means that more neural substrates are obtained from cerebral ischemia through the underlying neuroprotective mechanisms, which is beneficial for neurologic recovery after ischemic stroke [
20].
The knowledge of the many mechanisms, mediation of the inflammatory response, and promotion of angiogenesis, for preconditioning may reduce the neural deficits associated with ischemia and provide brain ischemic tolerance [
21]. These mechanisms are related with chronic intracerebral infection of
T. gondii in the host microenvironments.
T. gondii preferentially infects host monocyte/macrophage lineage cells, such as microglia [
22], and also contributes to the resolution of inflammation by releasing IL-10 and TGF-β (transforming growth factor-beta) through the differentiation of M2 phenotype macrophages [
13,
23–
25]. The evasion of anti-parasitic effector mechanisms as mediators of the inflammatory response by
T. gondii may be induced by brain ischemic tolerance [
26].
The HIF family as master regulatory transcription factors up-regulate multiple genes controlling neovascularization [
27]. Especially, HIF-1 is an important regulator for the transcription of VEGF, which is a major inducer of angiogenesis in hypoxic regions [
28]. Interestingly, HIF-1 was required for parasite cell division and organelle maintenance under hypoxic conditions. Specially,
T. gondii upregulates the expression of the HIF-1α subunit and activates HIF-1 reporter gene for growth and survival at physiologically relevant oxygen levels [
29]. In this study, we also created confirmed that chronic infection of
T. gondii in the brain induced the expression of HIF-1α and VEGF to stimulate angiogenesis under the hypoxia condition created by MCAO. The physiological alteration by manipulation of
T. gondii in the brain microenvironment may be help protect the brain tissue from ischemic damages by inducing brain ischemic tolerance.
Although the present findings demonstrated the beneficial effects of
T. gondii chronic infection on ischemia, there are several limitations. First, high mortality after MCAO surgery attenuated the statistical power of experimental results. Hypothermia after the surgery can be one cause of high mortality and can be reduced by animal temperature control [
30]. Unfortunately, our experiment protocol did not include this procedure. Lower mortality in the infection group (66% in the control group versus 49% in the infection group) can be an evidence of the neuroprotective effect of
T. gondii infection. However, further confirmation is necessary. Second, more complex behavioral tests are needed to verify the beneficial effects of
T. gondii infection. Cerebral ischemia results in motor weakness and sensory change, and also various neurologic deficits, such as cognitive deficit, aphasia, visuospatial neglect, and others. The CNS symptoms in
T. gondii infection can be diverse, and detailed behavioral tests are necessary to evaluate
T. gondii as a protective agent for ischemic stroke. Third, the mechanism of neuroprotective effect of
T. gondii infection remains unclear.
Despite these limitations, the overall findings of our study indicate that chronic T. gondii infection may be a promising preconditioning to help induce brain ischemic tolerance. The molecular mechanisms for this phenomenon must be investigated to confirm the protective effect of T. gondii infection on the outcome of cerebral ischemia.
Notes
-
CONFLICT OF INTEREST
We have no conflict of interest related to this work.
Supplementary Information
ACKNOWLEDGMENT
This study was supported by a grant no. 04-2016-0840 from the SNUH Research Fund.
Fig. 1Measurement of corrected infarction volume in Toxoplasma gondii intracerebral chronic infection 3 days after cerebral ischemia. (A) The brains were sectioned coronally, and cerebral infarctions were identified using triphenyl tetrazolium chloride staining (scale bar=1 cm). (B) Corrected infarct volume and (C) the ratio of the infarct volume to the volume of contralateral and (D) ipsilateral hemisphere were significantly reduced. Note: n=6 in the control, 9 in the infection groups; *P<0.05, compared to the control group by the t-test. Toxo, Toxoplasma gondii chronic intracerebral infection group.
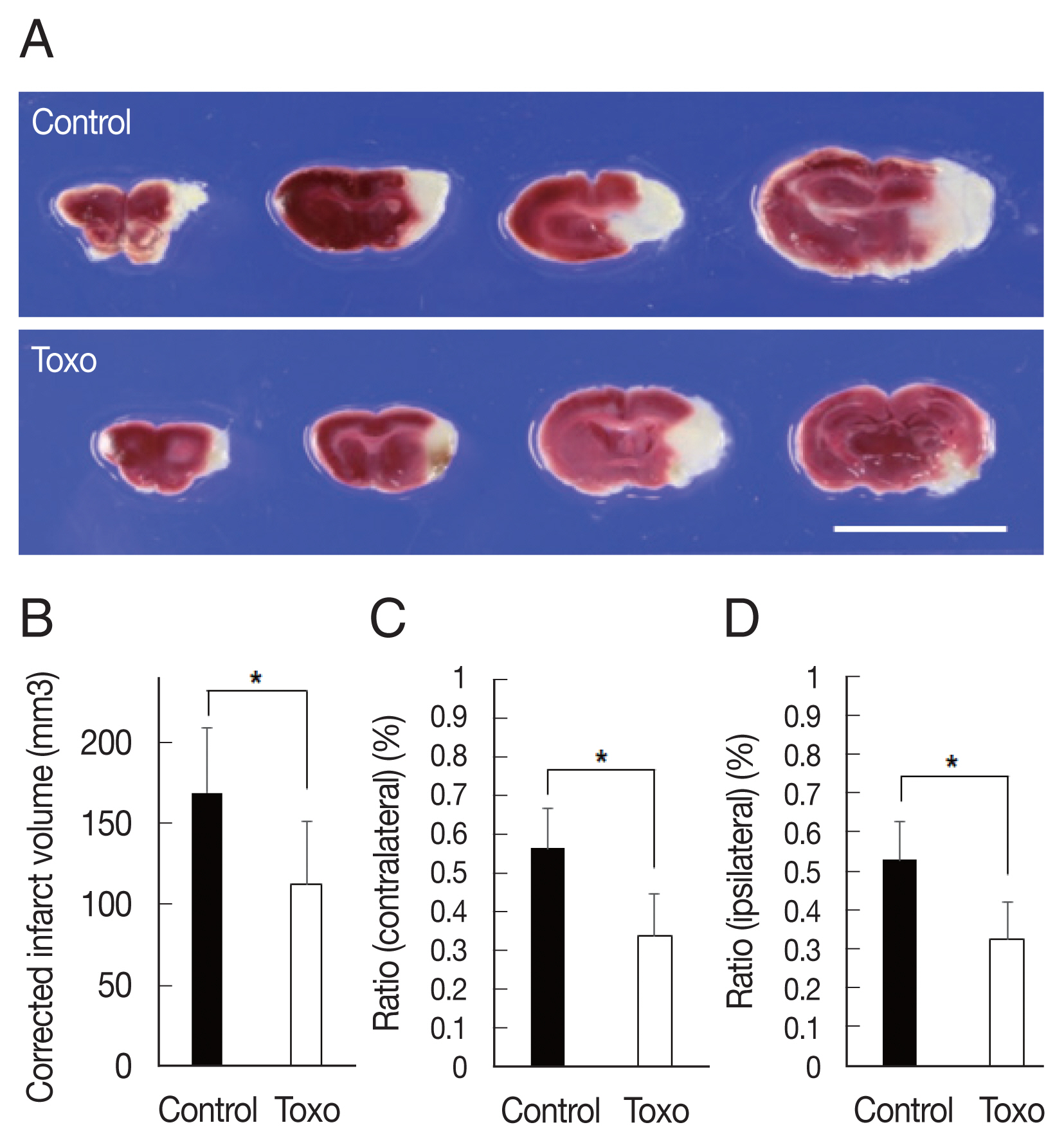
Fig. 2Effects of chronic intracerebral Toxoplasma gondii infection in neurologic deterioration after cerebral ischemia. Garcia score (A) and Rotarod test (B) were evaluated at baseline, day 1, and day 3 after middle cerebral artery occlusion. Note: n=6 in the control, 9 in the infection groups. *P< 0.05, compared to the control group by the t-test. Toxo, Toxoplasma gondii intracerebral chronic infection group.
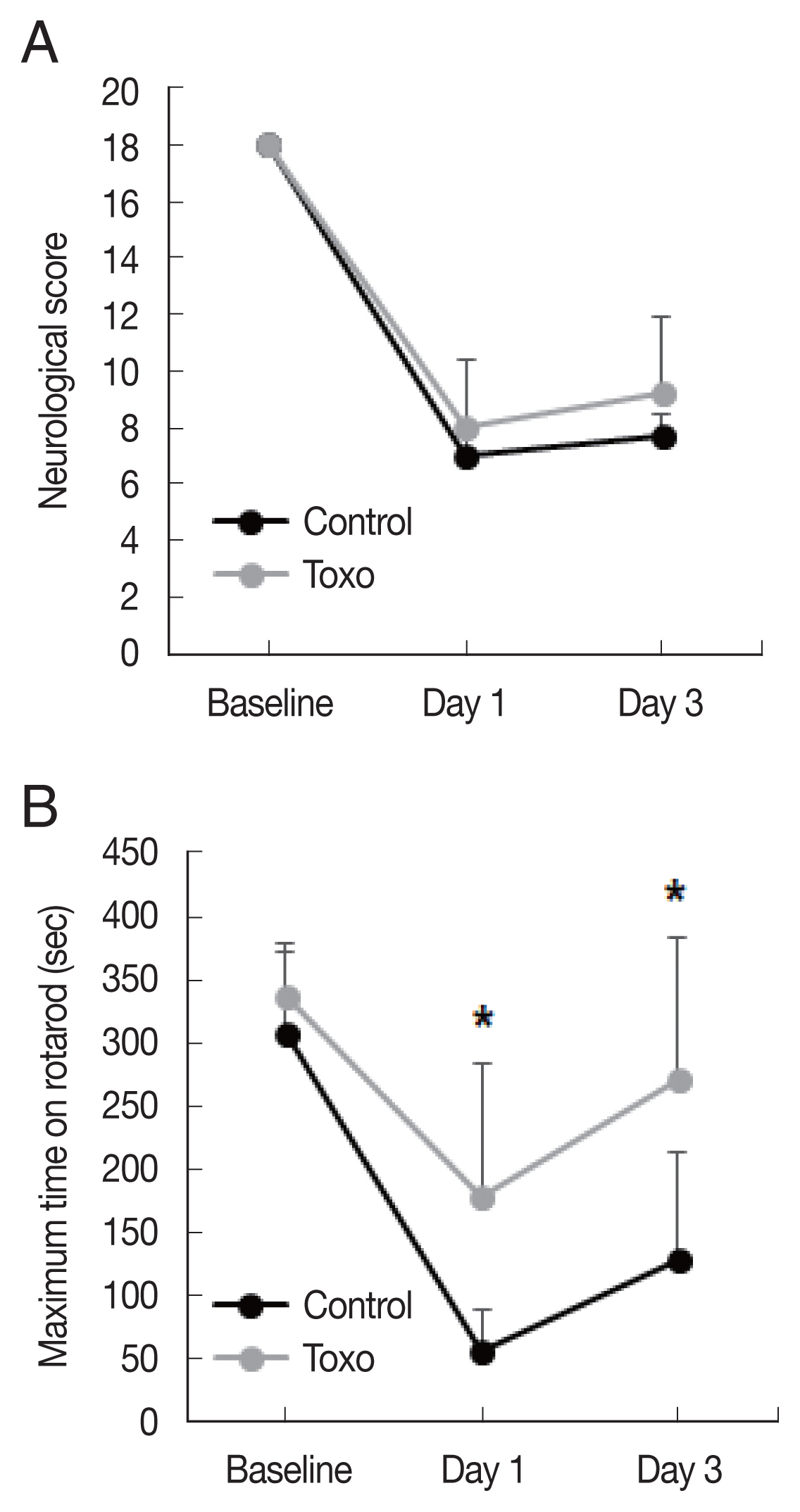
Fig. 3Expression levels of HIF-1α (A) and VEGF (B) in the brain of experimental mice infected with T. gondii after cerebral ischemia as determined using qRT-PCR and (C) western blotting. *P< 0.05, compared to the control group by the t-test. Con, Control; Toxo, Toxoplasma gondii intracerebral chronic infection group; HIF, hypoxia inducible factor; VEGF, vascular endothelial growth factor; MCAO, middle cerebral artery occlusion.
References
- 1. Jung BK, Song H, Lee SE, Kim MJ, Cho J, Shin EH, Chai JY. Seroprevalence and risk factors of Toxoplasma gondii infection among cat sitters in Korea. Korean J Parasitol 2017;55:203-206.
- 2. Jones JL, Dubey JP. Foodborne toxoplasmosis. Clin Infect Dis 2012;55:845-851.
- 3. Lindsay DS, Dubey JP.
Toxoplasma gondii: the changing paradigm of congenital toxoplasmosis. Parasitology 2011;138:1829-1831.
- 4. Bhopale GM. Pathogenesis of toxoplasmosis. Comp Immunol Microbiol Infect Dis 2003;26:213-222.
- 5. Dubey JP. Tissue cyst tropism in Toxoplasma gondii: a comparison of tissue cyst formation in organs of cats, and rodents fed oocysts. Parasitology 1997;115:15-20.
- 6. Parlog A, Schluter D, Dunay IR.
Toxoplasma gondii-induced neuronal alterations. Parasite Immunol 2015;37:159-170.
- 7. Hwang YS, Shin JH, Yang JP, Jung BK, Lee SH, Shin EH. Characteristics of infection immunity regulated by Toxoplasma gondii to maintain chronic infection in the brain. Front Immunol 2018;5:158.
- 8. Melzer TC, Cranston HJ, Weiss LM, Halonen SK. Host cell preference of Toxoplasma gondii cysts in murine brain: a confocal study. J Neuroparasitology 2010;1:N100505.
- 9. Vyas A, Kim SK, Giacomini N, Boothroyd JC, Sapolsky RM. Behavioral changes induced by Toxoplasma infection of rodents are highly specific to aversion of cat odors. Proc Natl Acad Sci U S A 2007;104:6442-6447.
- 10. Hermes G, Ajioka JW, Kelly KA, Mui E, Roberts F, Kasza K, Mayr T, Kirisits MJ, Wollmann R, Ferguson DJ, Roberts CW, Hwang JH, Trendler T, Kennan RP, Suzuki Y, Reardon C, Hickey WF, Chen L, McLeod R. Neurological and behavioral abnormalities, ventricular dilatation, altered cellular functions, inflammation, and neuronal injury in brains of mice due to common, persistent, parasitic infection. J Neuroinflammation 2008;5:48.
- 11. Schain M, Kreisl WC. Neuroinflammation in neurodegenerative disorders-a review. Curr Neurol Neurosci Rep 2017;17:25.
- 12. Macrez R, Ali C, Toutirais O, Le Mauff B, Defer G, Dirnagl U, Vivien D. Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol 2011;10:471-480.
- 13. Jung BK, Pyo KH, Shin KY, Hwang YS, Lim H, Lee SJ, Moon JH, Lee SH, Suh YH, Chai JY, Shin EH.
Toxoplasma gondii infection in the brain inhibits neuronal degeneration and learning and memory impairments in a murine model of Alzheimer's disease. PLoS One 2012;7:e33312.
- 14. Arsenijevic D, de Bilbao F, Vallet P, Hemphill A, Gottstein B, Richard D, Giannakopoulos P, Langhans W. Decreased infarct size after focal cerebral ischemia in mice chronically infected with Toxoplasma gondii
. Neuroscience 2007;150:537-546.
- 15. Morris GP, Wright AL, Tan RP, Gladbach A, Ittner LM, Vissel B. A comparative study of variables influencing ischemic injury in the longa and koizumi methods of intraluminal filament middle cerebral artery occlusion in mice. PLoS One 2016;11:e0148503.
- 16. Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 1995;26:627-634. discussion 635.
- 17. Rogers DC, Campbell CA, Stretton JL, Mackay KB. Correlation between motor impairment and infarct volume after permanent and transient middle cerebral artery occlusion in the rat. Stroke 1997;28:2060-2065. discussion 2066.
- 18. Bederson JB, Pitts LH, Germano SM, Nishimura MC, Davis RL, Bartkowski HM. Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke 1986;17:1304-1308.
- 19. Belayev L, Khoutorova L, Deisher TA, Belayev A, Busto R, Zhang Y, Zhao W, Ginsberg MD. Neuroprotective effect of SolCD39, a novel platelet aggregation inhibitor, on transient middle cerebral artery occlusion in rats. Stroke 2003;34:758-763.
- 20. Nelles G, Jentzen W, Bockisch A, Diener HC. Neural substrates of good and poor recovery after hemiplegic stroke: a serial PET study. J Neurol 2011;258:2168-2175.
- 21. Zhang F, Wu Y, Jia J. Exercise preconditioning and brain ischemic tolerance. Neuroscience 2011;177:170-176.
- 22. Butcher BA, Fox BA, Rommereim LM, Kim SG, Maurer KJ, Yarovinsky F, Herbert DR, Bzik DJ, Denkers EY.
Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog 2011;7:e1002236.
- 23. Albina JE, Mills CD, Henry WL Jr, Caldwell MD. Temporal expression of different pathways of 1-arginine metabolism in healing wounds. J Immunol 1990;144:3877-3880.
- 24. Kreider T, Anthony RM, Urban JF Jr, Gause WC. Alternatively activated macrophages in helminth infections. Curr Opin Immunol 2007;19:448-453.
- 25. Raes G, Beschin A, Ghassabeh GH, De Baetselier P. Alternatively activated macrophages in protozoan infections. Curr Opin Immunol 2007;19:454-459.
- 26. Sanad MM, Al Olayan EM.
Toxoplasma gondii strategy for intracellular survival: is it still enigmatic? Res J Parasitol 2011;6:60-73.
- 27. Oladipupo S, Hu S, Kovalski J, Yao J, Santeford A, Sohn RE, Shohet R, Maslov K, Wang LV, Arbeit JM. VEGF is essential for hypoxia-inducible factor-mediated neovascularization but dispensable for endothelial sprouting. Proc Natl Acad Sci U S A 2011;108:13264-13269.
- 28. Ziello JE, Jovin IS, Huang Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med 2007;80:51-60.
- 29. Spear W, Chan D, Coppens I, Johnson RS, Giaccia A, Blader IJ. The host cell transcription factor hypoxia-inducible factor 1 is required for Toxoplasma gondii growth and survival at physiological oxygen levels. Cell Microbiol 2006;8:339-352.
- 30. Wang-Fischer Y. Manual of Stroke Models in Rats. 1st ed. Boca Raton, USA. CRC Press; 2008.